
La toxicologie est l’étude des substances toxiques et, plus précisément, l’identification et l’évaluation quantitative des conséquences néfastes liées à l’exposition à des agents physiques, chimiques ou de toute autre nature. Comme telle, elle fait appel, tant pour ses connaissances que pour sa démarche de recherche ou ses méthodes, à la plupart des sciences biologiques fondamentales, aux disciplines médicales, à l’épidémiologie et à divers domaines de la chimie et de la physique. Elle s’étend de la recherche fondamentale sur le mécanisme d’action des agents toxiques à la mise au point et à l’interprétation de tests normalisés permettant de caractériser les propriétés toxiques de ces agents. Elle fournit à la médecine et à l’épidémiologie des informations indispensables pour comprendre l’étiologie et établir le lien entre les expositions, y compris professionnelles, et les pathologies observées. La toxicologie peut être scindée en spécialités: toxicologie clinique, toxicologie médico-légale, toxicologie fondamentale et toxicologie réglementaire, être présentée selon les organes cibles (par exemple, immunotoxicologie, toxicogénétique) ou encore selon ses objectifs (recherche, expérimentation et évaluation du risque).
Vouloir présenter de façon exhaustive la toxicologie dans cette Encyclopédie relève de la gageure. Ce chapitre ne saurait tenir lieu ni d’aide-mémoire sur cette discipline ni d’abrégé des connaissances sur les effets nocifs des divers agents toxiques. Ces informations sont plutôt à rechercher dans les bases de données continuellement mises à jour, comme nous l’expliquons dans la dernière partie du présent chapitre. Il ne tente pas non plus d’aborder des branches particulières de la toxicologie telle que la toxicologie médico-légale, mais bien de fournir des informations utilisables dans les différentes activités de cette discipline, ainsi que dans divers domaines médicaux et spécialités. Les thèmes ont été choisis en raison de leur orientation délibérément pratique et pour faciliter le renvoi à d’autres références au sein de la présente Encyclopédie.
Dans les sociétés modernes, la toxicologie est devenue un élément important pour assurer la santé tant dans le domaine environnemental que professionnel. C’est pourquoi de nombreuses organisations gouvernementales et non gouvernementales font appel à son fonds de connaissances pour évaluer les risques en milieu professionnel ou dans l’environnement en général et proposer une réglementation. Partie intégrante des stratégies de prévention, la toxicologie est d’une valeur inestimable, puisqu’elle est la source d’informations sur les risques potentiels en l’absence d’expositions humaines pertinentes. Il faut aussi rappeler que l’industrie emploie beaucoup les méthodes toxicologiques puisqu’elle y puise des renseignements utiles à la formulation de nouveaux produits ou à la conception de nouvelles molécules.
Le présent chapitre commence par cinq articles sur les principes généraux de la toxicologie, importants pour aborder la plupart des thèmes. Le premier concerne les relations entre exposition externe et dose interne. Dans la terminologie moderne, l’«exposition» fait référence aux concentrations ou quantités d’une substance auxquelles sont soumis des individus ou une population — quantités trouvées dans un volume donné d’air, d’eau ou de sol. La «dose» représente la concentration ou la quantité d’une substance présente chez un individu ou dans un organisme exposé. En santé au travail, les valeurs de référence et les lignes directrices sont souvent établies en terme d’exposition ou de limites admissibles de concentrations dans des situations spécifiques, par exemple dans l’atmosphère du lieu de travail. Ces limites d’exposition sont fondées sur la connaissance, ou parfois les hypothèses, de relations existant entre l’exposition et la dose; cependant, la dose interne n’étant pas toujours connue, de nombreuses études de santé au travail se limitent à déduire une association entre l’exposition et la réponse ou l’effet. Dans quelques cas, les normes ont été établies d’après la dose (par exemple, pour les taux admissibles de plomb dans le sang ou de mercure dans l’urine). Bien que ces mesures soient directement corrélées à la toxicité, il est encore nécessaire de recalculer les taux d’exposition correspondant à ces valeurs afin de mieux maîtriser les risques.
L’article qui suit, «Définitions et concepts», a trait aux facteurs et aux événements déterminant les relations entre exposition, dose et réponse. Les premiers de ces facteurs concernent la captation tissulaire, l’absorption et la distribution — processus qui déterminent le transport réel des substances dans le corps depuis l’environnement externe à travers les portes d’entrée telles que la peau, les poumons ou l’intestin. Ces processus sont à l’interface entre l’être humain et son environnement. Les seconds facteurs, métaboliques, objectivent la façon dont l’organisme traite les substances absorbées. Certaines d’entre elles sont transformées par les processus cellulaires du métabolisme qui peut soit renforcer leur activité biologique, soit l’atténuer.
Les concepts d’organe cible et d’effet critique ont été élaborés pour faciliter l’interprétation des données toxicologiques. Selon la dose, la durée et la voie d’exposition, et selon des facteurs intrinsèques tels que l’âge, de nombreux agents toxiques peuvent induire des effets divers au niveau des organes et des organismes. Un des principaux rôles de la toxicologie est de déterminer l’effet ou la série d’effets importants afin de prévenir l’apparition de maladies irréversibles ou invalidantes. Pour cela, il convient surtout d’identifier l’organe touché en premier ou le plus affecté par l’agent toxique: cet organe est appelé «organe cible». A l’intérieur de cet organe, il est capital de déceler le ou les événements importants objectivant une intoxication ou une lésion et permettant de mettre en évidence une altération de l’organe. Ce premier événement d’une série d’étapes physiopathologiques (l’excrétion de protéines de faible poids moléculaire représente un effet critique en néphrotoxicité), ou le premier effet potentiellement irréversible dans le processus d’une maladie (la formation d’adduits à l’ADN lors du processus de cancérogenèse), est appelé «l’effet critique». Ces concepts sont importants en santé au travail, car ils permettent de préciser le type de toxicité et de pathologie associé à une exposition spécifique; dans la plupart des cas, la diminution de l’exposition aura pour unique objectif de prévenir les effets critiques au niveau des organes cibles, et non ceux de l’ensemble des effets observés dans les différents organes.
Les deux articles suivants concernent les effets intrinsèques qui modifient les réponses à de nombreux agents toxiques. Il s’agit des déterminants génétiques, des facteurs de sensibilité ou de résistance héréditaire, de l’âge, du sexe ou encore de paramètres tels que le régime alimentaire ou la coexistence d’une maladie infectieuse. Ces facteurs peuvent aussi affecter l’exposition et la dose, en modifiant la captation tissulaire, l’absorption, la distribution et le métabolisme. Etant donné la diversité de ces facteurs parmi les travailleurs dans le monde, il est essentiel que les spécialistes de la santé au travail et les décideurs comprennent la façon dont ils font varier les réponses en fonction de la population et des individus à l’intérieur de la population. Dans les sociétés qui ont une population hétérogène, cette question est particulièrement importante. Cette disparité des populations humaines doit être prise en compte pour évaluer les risques d’exposition professionnelle et tirer des conclusions rationnelles à partir d’études expérimentales effectuées dans le cadre des recherches toxicologiques.
Cette section aborde ensuite deux aspects généraux des mécanismes d’action en toxicologie. De ce point de vue, les toxicologues modernes considèrent que tous les effets toxiques s’exercent en premier lieu au niveau cellulaire; les réponses cellulaires représentent donc les signes les plus précoces de la lutte de l’organisme vis-à-vis d’un agent toxique et font probablement partie d’une suite d’événements qui va de la lésion initiale jusqu’à la mort cellulaire. La lésion cellulaire fait appel à des processus spécifiques auxquels la cellule, plus petite unité d’organisation biologique dans un organe, a recours pour répondre à l’atteinte. Ces réponses impliquent des modifications dans le fonctionnement des processus cellulaires, en particulier au niveau de la membrane dont on connaît les rôles d’absorption, de sécrétion et d’excrétion des substances, de la synthèse protéique à partir d’acides aminés et du renouvellement des composants cellulaires. Ces réponses peuvent être communes à toutes les cellules endommagées, ou être spécifiques à des types cellulaires particuliers de certains organes. La mort cellulaire est la destruction des cellules dans un système organique, par suite d’une lésion cellulaire irréversible ou non compensée. Elle peut survenir lors d’une intoxication aiguë, comme dans le cas des agents toxiques agissant sur le transfert d’oxygène, ou être la conséquence d’une intoxication chronique. Elle peut être suivie d’une régénération dans certains organes, bien que cette prolifération puisse, dans certaines conditions, être considérée comme une réponse toxique. Même en l’absence de mort cellulaire, une lésion répétée peut induire un stress au niveau d’un organe susceptible d’altérer ses fonctions et son devenir.
Le présent chapitre traite ensuite de domaines plus spécifiques, regroupés selon les catégories suivantes: mécanismes, méthodologies, réglementation et évaluation du risque. Les articles portant sur les mécanismes mettent l’accent principalement sur les systèmes cibles plutôt que sur les organes. Cette présentation est à l’image de la pratique de la médecine et de la toxicologie modernes, qui étudient les systèmes plutôt que les organes isolés. Ainsi, les commentaires sur la toxicogénétique ne concernent pas uniquement les effets toxiques des agents sur un organe spécifique, mais bien le matériel génétique en tant que cible de l’action toxique. De même, l’article sur l’immunotoxicologie examine les divers organes et cellules du système immunitaire en tant que cibles vis-à-vis des agents toxiques. Les articles méthodologiques se veulent avant tout opérationnels; ils décrivent les méthodes qu’on emploie actuellement dans de nombreux pays pour identifier les risques, ou la manière dont on élabore l’information sur les propriétés biologiques des agents toxiques.
Le chapitre se poursuit par cinq articles relatifs à l’application de la toxicologie sur le plan réglementaire et décisionnel, depuis l’identification du risque jusqu’à son évaluation. Les procédures actuellement suivies dans différents pays y sont présentées, de même que celles du Centre international de recherche sur le cancer (CIRC). Ces articles devraient permettre au lecteur de comprendre comment, à partir de l’information tirée des tests toxicologiques associés à des déductions mécanistiques et fondamentales, l’on parvient à une information quantitative qui sert ensuite à établir les niveaux d’exposition et à d’autres approches permettant de maîtriser les risques sur le lieu de travail et dans l’environnement en général.
D’autres chapitres de la présente Encyclopédie renseignent sur les bases de données concernant la toxicologie. Ces bases fournissent aux spécialistes de la santé au travail, aux travailleurs et aux employeurs une information actuelle sur la toxicologie et l’évaluation des agents toxiques par des organismes nationaux et internationaux.
Le présent chapitre porte sur la toxicologie dans ses relations avec la sécurité et la santé au travail. Pour cette raison, la toxicologie clinique et la toxicologie médico-légale n’y sont pas expressément abordées. De nombreux principes et de nombreuses démarches semblables à ceux qui y sont décrits sont utilisés dans ces sous-disciplines de la même façon qu’en santé environnementale. Ils sont également applicables pour évaluer l’impact des agents toxiques sur les populations autres qu’humaines, préoccupation majeure des politiques environnementales dans de nombreux pays. On a tenté, dans ce chapitre, de présenter les points de vue et les expériences des experts et des praticiens de tous les secteurs dans de nombreux pays; cependant, le lecteur pourra noter un certain parti pris envers les scientifiques universitaires du monde développé. Bien que l’éditeur et ses collaborateurs soient convaincus que les principes et la pratique de la toxicologie sont internationaux, les préjugés culturels et le caractère restreint de l’expérience pourront paraître évidents dans ce chapitre. L’éditeur espère que les lecteurs de cette Encyclopédie profiteront de ses mises à jour pour conférer à cet ouvrage la perspective le plus large possible et contribuer à son enrichissement permanent.
La toxicité est la capacité intrinsèque d’un agent chimique à avoir un effet nocif sur un organisme.
Le terme xénobiotique désigne une «substance étrangère», c’est-à-dire extérieure à l’organisme, par opposition aux composants endogènes. Les xénobiotiques comprennent les médicaments, les produits chimiques industriels, les poisons naturels et les polluants environnementaux.
Un danger représente une toxicité potentielle pouvant survenir dans un cadre ou une situation déterminés.
Un risque est la probabilité d’apparition d’un effet nocif spécifique. Il est souvent exprimé en pourcentage de cas dans une population donnée pour une durée déterminée. Une évaluation du risque peut être faite à partir de cas réels ou par projection de cas futurs, basée sur des extrapolations.
L’évaluation de la toxicité, de même que la classification de la toxicité, peuvent être utilisées dans un but réglementaire. Il s’agit d’une classification arbitraire des doses ou des niveaux d’exposition («très toxique», «extrêmement toxique», «modérément toxique», etc.) à l’origine d’effets toxiques qui permet de répertorier les produits exerçant une toxicité aiguë. La classification de la toxicité permet de regrouper les produits chimiques dans des catégories générales selon leur effet toxique essentiel, par exemple les allergènes, les neurotoxiques, les cancérogènes, etc. Elle peut avoir une valeur administrative d’avertissement et d’information.
La relation dose-effet est la relation entre la dose et l’effet à l’échelle de l’individu. L’augmentation de la dose peut accroître l’intensité ou la sévérité d’un effet. Une courbe dose-effet peut être tracée pour l’ensemble de l’organisme, la cellule ou la molécule cible. Certains effets toxiques, comme la mort ou le développement d’un cancer, n’ont pas un caractère progressif: ils représentent des effets «tout ou rien».
La relation dose-réponse désigne la relation entre la dose et le pourcentage d’individus présentant un effet spécifique. Lorsque la dose augmente, un plus grand nombre d’individus sont affectés dans la population exposée.
Il est essentiel pour la toxicologie d’établir les relations dose-effet et dose-réponse. En médecine (épidémiologie), le critère de relation causale souvent employé entre un agent et une pathologie repose sur la proportionnalité entre la dose et les effets ou réponses observés.
Plusieurs courbes dose-réponse peuvent être tracées pour un même produit chimique — une par type d’effet. La courbe dose-réponse pour la plupart des effets toxiques (quand ils sont étudiés dans une population importante) a une forme sigmoïde. On observe généralement une zone de doses faibles où aucune réponse ne peut être détectée; avec l’augmentation de la dose, la réponse suit une courbe ascendante pour atteindre généralement un plateau à 100% de réponses. La courbe dose-réponse reflète les variations interindividuelles dans une population. La pente de la courbe varie d’un produit chimique à l’autre et selon le type d’effet. Dans le cas de certains produits chimiques présentant des effets spécifiques (cancérogènes, initiateurs, mutagènes), la courbe dose-réponse peut être linéaire pour une gamme de doses donnée dès la dose zéro. Cela signifie qu’il n’existe aucun seuil pour ces substances et que des doses mêmes faibles font encourir un risque. Au-delà de cette gamme de dose, le risque peut passer à un taux plus important que le taux linéaire.
Les variations d’exposition en cours de journée ou la durée totale d’exposition au cours d’une vie peuvent être aussi importantes pour le résultat observé que la dose moyenne ou même la dose intégrée. Des pics élevés d’exposition peuvent être plus dangereux qu’une exposition plus régulière. C’est le cas avec certains solvants organiques. Par ailleurs, pour certains cancérogènes, il a été démontré expérimentalement qu’à dose totale identique, le fractionnement en plusieurs expositions a une incidence accrue sur l’apparition de tumeurs.
La dose est souvent exprimée en tant que quantité (mg/kg de poids corporel) de xénobiotique ayant pénétré l’organisme. Elle peut être exprimée de différentes manières (plus ou moins informatives): dose d’exposition, concentration dans l’air d’un polluant inhalé durant une certaine période (huit heures en général en hygiène du travail); dose retenue ou absorbée (également appelée en hygiène du travail charge corporelle) qui est la quantité présente dans l’organisme à un moment donné pendant ou après une exposition. La dose tissulaire est la quantité de substance dans un tissu spécifique et la dose cible est la quantité de substance (généralement un métabolite) liée à la molécule critique. La dose cible est la quantité de produit chimique (en mg) fixée par mg de macromolécule spécifique dans un tissu. Pour utiliser ce concept, il faut disposer d’informations sur le mécanisme d’action au niveau moléculaire. La dose cible est associée plus précisément à l’effet toxique. La dose d’exposition ou la charge corporelle, plus facilement disponibles, sont liées de manière moins précise à l’effet toxique.
La notion de dose comporte souvent un paramètre temporel, même s’il n’est pas toujours exprimé. La dose théorique selon la loi de Haber est D = ct, où D est la dose, c la concentration du xénobiotique dans l’air et t la durée d’exposition à un produit chimique. Au niveau de l’organe cible ou au niveau moléculaire, on peut dire qu’il s’agit de la quantité fixée par mg de tissu ou de molécule pour un temps donné. La prise en compte du temps est généralement plus importante pour comprendre les expositions répétées et les effets chroniques que pour les expositions uniques et les effets aigus.
Les effets additifs sont le résultat d’une exposition combinée à plusieurs produits chimiques, où les toxicités particulières sont simplement additionnées les unes aux autres (1+1 = 2). Lorsque les produits chimiques agissent selon le même mécanisme, on peut présumer qu’ils auront un effet additif, mais il n’en va pas toujours de même dans la réalité. Ainsi, il peut arriver que l’interaction entre des produits chimiques aboutisse à une inhibition (antagonisme), l’effet observé étant plus faible que celui attendu par addition des effets des produits chimiques individuels (1+1<2). Inversement, la combinaison de produits chimiques peut produire un effet plus prononcé que celui attendu par simple addition (réponse augmentée chez les individus ou augmentation de la fréquence des réponses parmi une population) (synergie) (1+1>2).
Le temps de latence est le temps qui s’écoule entre une première exposition et l’apparition d’un effet ou d’une réponse décelables. Ce terme est souvent employé pour les effets cancérogènes, où les tumeurs apparaissent longtemps après le début de l’exposition et quelquefois bien après son arrêt.
Une dose seuil est le niveau de dose en dessous duquel aucun effet observable ne survient. Il existe des seuils pour certains effets, notamment les effets toxiques aigus, mais non pour d’autres, par exemple pour les effets cancérogènes (initiateurs formant des adduits à l’ADN). Une simple absence de réponse dans une population donnée ne saurait cependant être interprétée comme la preuve de l’existence d’un seuil. Elle peut être due à un simple phénomène statistique: un effet toxique ne se produisant qu’à faible fréquence pourra ne pas être décelé dans une petite population.
La DL50 (dose létale 50) est la dose qui entraîne le décès de la moitié du lot d’animaux de laboratoire soumis au toxique étudié. Elle est souvent employée dans la littérature classique comme une mesure de la toxicité aiguë des produits chimiques. Plus la DL50 est élevée, plus la toxicité aiguë est faible. Un produit chimique très toxique (avec une faible DL50) est dit violent. Il n’existe pas nécessairement de corrélation entre la toxicité aiguë et la toxicité chronique. La DE50 (dose efficace) est la dose responsable d’un effet spécifique autre que la létalité chez 50% des animaux.
La valeur NOEL (NOAEL) (No Observed (Adverse) Effect Level) correspond à la dose à laquelle aucun effet (nocif) n’est observé, ou encore la plus forte dose n’entraînant aucun effet toxique. Pour établir une valeur NOEL, il faut disposer de nombreuses doses dans une population importante mais aussi d’autres informations pour s’assurer que l’absence de réponse n’est pas simplement le résultat d’un phénomène statistique. La valeur LOEL (Low Observed Effect Level) correspond à la dose efficace la plus faible sur une courbe dose-réponse, ou à la plus faible dose provoquant un effet.
Un facteur de sécurité est un chiffre formel et arbitraire par lequel on divise les valeurs NOEL ou LOEL obtenues expérimentalement pour définir une dose admissible chez l’humain. Ce facteur, souvent employé en toxicologie alimentaire mais aussi en toxicologie professionnelle, peut servir à extrapoler des données issues de petites populations à des populations plus importantes. Les facteurs de sécurité varient de 100 à 103. On considère qu’un facteur de sécurité de deux suffit à protéger d’un effet peu sévère (par exemple, une irritation), alors que pour tous les effets très sévères (par exemple, un cancer), on applique un facteur pouvant aller jusqu’à 1 000. L’expression facteur de sécurité pourrait fort bien être remplacée par celle de facteur de protection ou, encore, facteur d’incertitude, notion qui reflète en effet mieux l’incertitude scientifique quant à savoir si des données dose-réponse concernant un produit chimique particulier, un effet toxique ou une condition d’exposition peuvent être extrapolées de l’animal à l’espèce humaine.
Les extrapolations sont des estimations théoriques qualitatives ou quantitatives de toxicité (extrapolation d’un risque) obtenues par déduction de données d’une espèce à l’autre, ou d’un ensemble de données dose-réponse obtenues dans une zone de doses élevées à des zones de dose-réponse pour lesquelles il n’existe pas de données. Elles permettent de prévoir une réponse toxique en dehors du champ d’observation. On les établit à partir de modèles mathématiques basés sur la connaissance du devenir d’un produit chimique dans l’organisme (modèle toxicocinétique) ou sur la probabilité statistique de la survenue d’un mécanisme biologique (modèle biologique ou mécanistique). Certains organismes nationaux ont mis au point, dans un but réglementaire, des modèles d’extrapolation complexes permettant de prévoir un risque (voir commentaires sur l’évaluation du risque plus loin dans ce chapitre).
Les effets systémiques sont les effets toxiques observés dans des tissus éloignés de la voie d’absorption.
L’organe cible est l’organe principal ou l’organe le plus sensible atteint lors d’une exposition. Un même produit chimique pénétrant dans l’organisme peut atteindre des organes cibles différents selon la voie, la dose, le sexe et l’espèce. Une interaction entre produits chimiques, ou entre produits chimiques et d’autres facteurs, peut également affecter différents organes cibles.
Les effets aigus sont des effets survenant rapidement (en général en moins de vingt-quatre heures) après une exposition limitée; ils peuvent être réversibles ou irréversibles.
Les effets chroniques surviennent après une exposition prolongée (mois, années, décennies) ou persistent une fois que l’exposition a cessé.
Une exposition aiguë est une exposition de courte durée, tandis qu’une exposition chronique est une exposition de longue durée (parfois toute la vie).
La tolérance (appelée aussi accoutumance ou mithridatisation) est le phénomène qui se produit lorsque des expositions répétées entraînent une réponse inférieure à celle que l’on observe sans prétraitement.
Diffusion. Pour pénétrer dans l’organisme et atteindre le site où elle exercera sa toxicité, une substance étrangère doit franchir plusieurs obstacles, y compris les cellules et leurs membranes. La plupart des substances toxiques traversent les membranes passivement par diffusion. Ainsi, les petites molécules hydrosolubles passent à travers les canaux aqueux, les molécules liposolubles pénétrant par dissolution et diffusion à travers la partie lipidique de la membrane. L’éthanol, petite molécule à la fois hydro- et liposoluble, diffuse rapidement à travers les membranes cellulaires.
Diffusion des acides et bases faibles. Les acides et bases faibles peuvent facilement traverser les membranes sous leur forme non ionisée liposoluble, alors que les formes ionisées trop polaires ne le peuvent pas. Le degré d’ionisation de ces substances dépend du pH. S’il existe un gradient de pH de part et d’autre d’une membrane, elles s’accumuleront d’un seul côté. L’excrétion urinaire des acides et des bases faibles est fortement dépendante du pH urinaire. Le pH fœtal ou embryonnaire est un peu plus élevé que le pH maternel, ce qui explique la tendance des acides faibles à s’accumuler dans le fœtus ou l’embryon.
Diffusion facilitée. Le passage d’une substance peut être facilité par l’existence de transporteurs membranaires. La diffusion facilitée est comparable à un processus enzymatique dans la mesure où elle est sous la dépendance d’une protéine fortement sélective et saturable. D’autres substances peuvent inhiber le transport facilité des xénobiotiques.
Transport actif. Certaines substances sont activement transportées à travers les membranes cellulaires. Ce transport s’effectue par l’intermédaire de protéines porteuses selon un processus analogue à celui des enzymes. Le transport actif s’apparente à la diffusion facilitée, mais il peut se produire contre un gradient de concentration. Il requiert un apport d’énergie et peut être bloqué par un inhibiteur métabolique. La plupart des polluants environnementaux ne sont pas transportés de manière active. La sécrétion et la réabsorption actives au niveau tubulaire rénal des métabolites acides constituent une exception.
Phagocytose. Il s’agit d’un processus par lequel des cellules spécialisées comme les macrophages absorbent des particules en vue de les dégrader. Ce processus de transport est important, par exemple pour l’élimination de particules au niveau des alvéoles pulmonaires.
Flux de masse. Les substances sont aussi transportées dans l’organisme avec le flux de l’air, ou par le flux sanguin, lymphatique ou urinaire.
Filtration. L’eau traverse les pores endothéliaux sous l’influence de la pression hydrostatique ou osmotique. Tout soluté de faible poids moléculaire sera filtré en même temps que l’eau. Une partie de la filtration se fait au niveau du lit capillaire dans tous les tissus; elle est particulièrement importante pour la formation de l’urine primaire dans les glomérules rénaux.
L’absorption est l’incorporation d’une substance par l’organisme. Ce terme comprend habituellement non seulement le passage à travers la barrière tissulaire, mais aussi le transport ultérieur vers la circulation sanguine.
Absorption pulmonaire. Les poumons constituent la voie essentielle de dépôt et d’absorption des petites particules aériennes, des gaz, des vapeurs et des aérosols. Dans le cas des gaz et des vapeurs très hydrosolubles, l’incorporation se fait pour l’essentiel au niveau du nez et de l’arbre respiratoire, alors que pour les substances moins hydrosolubles elle s’effectue surtout dans les alvéoles pulmonaires. Les alvéoles ont une surface très importante (environ 100 m2 chez l’humain). De plus, la barrière de diffusion est extrêmement mince puisqu’elle est constituée de deux couches cellulaires fines formant un espace de l’ordre de quelques microns entre l’air alvéolaire et la circulation sanguine systémique. Les poumons sont donc très efficaces non seulement pour les échanges oxygène et gaz carbonique, mais aussi pour les autres gaz et vapeurs. En général, la diffusion à travers la paroi alvéolaire est si rapide qu’elle ne limite pas le transport. Le taux d’absorption dépend par contre du flux (ventilation pulmonaire, débit cardiaque) et de la solubilité (coefficient de partage sang:air). Un autre facteur important est l’élimination métabolique. L’importance relative de ces facteurs sur l’absorption pulmonaire varie beaucoup selon les substances. L’activité physique entraîne une augmentation de la ventilation pulmonaire et du débit cardiaque, ainsi qu’une diminution du flux sanguin hépatique (et, partant, du taux de biotransformation). Pour beaucoup de substances inhalées, cela se traduit par une augmentation marquée de l’absorption pulmonaire.
Absorption percutanée. La peau est une barrière très efficace. A côté de son rôle thermorégulateur, elle est conçue pour protéger l’organisme contre les micro-organismes, le rayonnement ultraviolet et autres agents nocifs et éviter une perte d’eau excessive. La distance de diffusion dans le derme est de l’ordre de quelques dixièmes de millimètres. De plus, la couche de kératine présente une très grande résistance à la diffusion pour la plupart des substances. Néanmoins, en présence de substances liposolubles très toxiques telles que les insecticides organophosphorés ou les solvants organiques, on peut observer une absorption dermique considérable pouvant être à l’origine d’une intoxication. Dans le cas de substances liquides, l’absorption est notable. L’absorption percutanée de vapeurs peut être importante pour les solvants présentant une pression de vapeur très basse et une forte affinité pour l’eau et la peau.
Absorption gastro-intestinale. Elle survient par ingestion accidentelle ou volontaire. Les plus grosses particules inhalées et déposées dans l’appareil respiratoire peuvent être avalées après transport mucociliaire vers le pharynx. En pratique, toutes les substances solubles sont efficacement absorbées dans l’appareil gastro-intestinal. Le pH acide de l’intestin facilite l’absorption de certains toxiques, les métaux par exemple.
Autres voies. En toxicologie expérimentale, on utilise pour des raisons de commodité d’autres voies d’administration, alors qu’elles sont rares et non pertinentes en milieu professionnel: les injections intraveineuses (iv), sous-cutanées (sc), intrapéritonéales (ip) et intramusculaires (im). D’une façon générale, ces voies parentérales permettent une absorption plus rapide et plus complète des substances, surtout dans le cas de la voie intraveineuse. On obtient alors des pics de concentration de courte durée, mais élevés, à l’origine d’une plus forte toxicité de la dose administrée.
La distribution d’une substance dans l’organisme est un processus dynamique dépendant des vitesses de captation tissulaire et d’élimination, du flux sanguin vers les différents tissus et de l’affinité de ces derniers pour la substance. Les petites molécules hydrosolubles, non ionisées, les cations monovalents et la plupart des anions diffusent facilement et finissent par se répartir de façon relativement régulière dans l’organisme.
Volume de distribution. Il s’agit de la quantité d’une substance dans l’organisme, divisée par la concentration sanguine, plasmatique ou sérique à un moment donné. Cette valeur n’a pas de sens en termes de volume physique, de nombreuses substances n’étant pas distribuées uniformément dans l’organisme. Un volume de distribution inférieur à 1 litre par kg de poids corporel indique une distribution préférentielle dans le sang (le sérum ou le plasma), alors qu’une valeur supérieure témoigne d’une prédilection pour les tissus périphériques, par exemple le tissu adipeux pour les substances liposolubles.
Accumulation. Ce terme désigne l’accumulation d’une substance dans un tissu ou un organe à une concentration supérieure à celle présente dans le sang ou le plasma. Il peut également faire référence à une accumulation progressive dans l’organisme au cours du temps. De nombreux xénobiotiques sont fortement liposolubles et ont tendance à s’accumuler dans le tissu adipeux, alors que d’autres présentent une affinité particulière pour le tissu osseux. C’est ainsi que le calcium osseux peut s’échanger avec des cations tels que le plomb, le strontium, le baryum ou le radium, et les groupes hydroxyles osseux s’échanger avec des ions fluorure.
Barrières. Les vaisseaux sanguins au niveau du cerveau, des testicules et du placenta présentent des structures anatomiques spéciales empêchant le passage des grosses molécules telles que les protéines. Ces structures, souvent appelées barrières hémato-méningée, testiculaire et placentaire, peuvent donner l’impression erronée qu’elles empêchent le passage de toute substance quand elles ont en fait peu d’importance, voire aucune, pour les xénobiotiques qui peuvent diffuser à travers les membranes cellulaires.
Liaison sanguine. Les substances peuvent être liées aux hématies, aux composants plasmatiques, ou se trouver à l’état libre non liées dans le sang. Le monoxyde de carbone, l’arsenic, le mercure organique et le chrome hexavalent ont une forte affinité pour les hématies, alors que le mercure inorganique et le chrome trivalent montrent une prédilection pour les protéines plasmatiques. De nombreuses autres substances sont également liées aux protéines plasmatiques. Seule la fraction libre est disponible pour la filtration et la diffusion vers les organes d’élimination. Ainsi, la liaison sanguine peut faire augmenter la durée du séjour dans l’organisme et diminuer la captation tissulaire au niveau des organes cibles.
L’élimination est la phase qui assure la disparition d’une substance de l’organisme soit parce qu’elle est excrétée, soit parce qu’elle est transformée en d’autres produits qui ne sont plus décelables. La vitesse de disparition peut être exprimée par la constante d’élimination, la demi-vie biologique ou la clairance.
La courbe temps-concentration. La courbe de concentration dans le sang (ou le plasma) en fonction du temps est une manière pratique de décrire la captation tissulaire et le devenir d’un xénobiotique.
L’aire sous la courbe taux plasmatique-temps est l’intégrale de la concentration dans le sang (ou le plasma) au cours du temps. En l’absence de saturation métabolique et d’autres processus non linéaires, l’aire sous la courbe est proportionnelle à la quantité absorbée de substance.
La demi-vie biologique (ou demi-vie) est le temps nécessaire après la fin d’une exposition pour réduire de moitié la quantité de substance présente dans l’organisme. Comme il est souvent difficile d’évaluer la quantité totale d’une substance, on mesure sa concentration sanguine (plasmatique). La demi-vie doit être utilisée avec précaution, car elle peut varier, par exemple, selon la dose et la durée d’exposition. De plus, de nombreuses substances ont des courbes de décroissance complexes avec plusieurs demi-vies.
La biodisponibilité est la fraction d’une dose administrée pénétrant dans la circulation systémique. En l’absence de clairance présystémique, ou de métabolisme de premier passage, la fraction est égale à 1. Lors d’une exposition per os, la clairance présystémique peut être due au métabolisme au niveau du contenu gastro-intestinal, de la paroi intestinale ou du foie. Le métabolisme de premier passage réduit l’absorption systémique de la substance et accroît plutôt l’absorption des métabolites, ce qui peut modifier le type de toxicité.
La clairance est le volume de sang (plasma) complètement épuré d’une substance par unité de temps; c’est aussi le rapport entre le débit urinaire, par minute, d’un corps et sa concentration dans le plasma. Afin de la distinguer de la clairance rénale, on parle de clairance totale, métabolique ou sanguine (plasmatique).
La clairance intrinsèque est l’aptitude des enzymes endogènes à transformer une substance; elle est également exprimée en volume par unité de temps. Si la clairance intrinsèque d’un organe est plus faible que le flux sanguin, le métabolisme est dit à capacité limitée. Inversement, si la clairance intrinsèque est beaucoup plus élevée que le flux sanguin, le métabolisme est limité par le flux.
L’excrétion est l’élimination de l’organisme d’une substance et de ses produits de biotransformation.
Excrétion dans l’urine et la bile. Les reins sont les organes excréteurs les plus importants. Certaines substances, en particulier les acides de poids moléculaire élevé, sont excrétées par la bile. Une fraction des substances ainsi excrétées peut être réabsorbée au niveau intestinal. Ce processus, appelé circulation entéro-hépatique, est habituel pour les substances conjuguées après hydrolyse intestinale.
Autres voies d’excrétion. Certaines substances, les solvants organiques, des produits de dégradation comme l’acétone, sont suffisamment volatiles pour qu’une fraction importante puisse être éliminée par exhalaison après leur inhalation. Les molécules hydrosolubles ou liposolubles de faible poids moléculaire sont facilement sécrétées vers le fœtus par voie placentaire, et dans le lait chez les mammifères. Chez la mère, la lactation peut être une voie d’excrétion importante du point de vue quantitatif pour les produits chimiques liposolubles. La descendance peut être secondairement exposée par l’intermédiaire de la mère pendant la grossesse et lors de la lactation. La sueur et la salive peuvent aussi servir d’émonctoire, bien que beaucoup moins important, aux composés hydrosolubles. Cependant, étant donné le volume de salive produit et absorbé, l’excrétion salivaire peut contribuer à la réabsorption d’un produit. Certains métaux, comme le mercure, sont excrétés dans les cheveux par suite de leur forte liaison aux groupes sulphydryles de la kératine.
Les modèles mathématiques sont des outils importants pour comprendre et décrire la captation tissulaire et la répartition des substances étrangères. La plupart des modèles sont compartimentaux, l’organisme étant représenté par un ou plusieurs compartiments. Un compartiment est un volume théorique du point de vue chimique et physique dans lequel la substance est censée se distribuer de manière homogène et instantanée. Les modèles simples sont exprimés comme une somme de termes exponentiels, alors que les plus complexes requièrent des calculs numériques sur ordinateur. Les modèles peuvent être subdivisés en deux catégories: descriptive et physiologique.
Dans les modèles descriptifs, on assure l’ajustement des données mesurées en modifiant les valeurs numériques des paramètres du modèle ou même la structure de celui-ci. La structure du modèle n’a normalement que peu de rapport avec celle de l’organisme. Ces modèles présentent l’avantage de ne nécessiter que peu d’hypothèses et aucune donnée supplémentaire; ils ont par contre l’inconvénient de n’avoir qu’une utilisation limitée pour les extrapolations.
Les modèles physiologiques sont construits à partir de données physiologiques indépendantes, anatomiques et autres. Le modèle est alors affiné et validé par comparaison avec les données expérimentales. Un des avantages des modèles physiologiques est qu’ils peuvent servir à faire des extrapolations. Par exemple, ils permettent de prédire l’influence de l’activité physique sur la captation tissulaire et la répartition des substances inhalées du fait des ajustements physiologiques connus de la ventilation et du débit cardiaque. Ces modèles requièrent malheureusement une quantité importante de données indépendantes.
La biotransformation est un processus qui mène à la transformation métabolique de composés étrangers (xénobiotiques) dans l’organisme. Ce processus est souvent appelé métabolisme des xénobiotiques. En règle générale, le métabolisme convertit les xénobiotiques liposolubles en métabolites hydrosolubles, de poids moléculaire plus élevé et faciles à éliminer.
Le foie est le principal site de la biotransformation. Tous les xénobiotiques absorbés au niveau intestinal sont transportés vers le foie par un vaisseau sanguin unique, la veine porte. Si une substance étrangère est absorbée en petites quantités, elle peut être complètement métabolisée par le foie avant d’atteindre la circulation générale et les autres organes (effet de premier passage). Les xénobiotiques inhalés parviennent au foie par la circulation générale. Seule une fraction de la dose est alors métabolisée avant d’atteindre les autres organes.
Les cellules hépatiques contiennent diverses enzymes qui oxydent les xénobiotiques. Cette oxydation active généralement le composé, qui devient plus réactif que la molécule mère. Dans la plupart des cas, le métabolite oxydé est métabolisé plus complètement par d’autres enzymes lors d’une seconde phase. Ces enzymes conjuguent le métabolite avec une substance endogène, de sorte que la molécule augmente de volume et se polarise, ce qui facilite son élimination.
Les enzymes métabolisant les xénobiotiques sont également présentes dans d’autres organes tels que les poumons et les reins où elles peuvent jouer des rôles spécifiques et qualitativement importants dans le métabolisme de certains xénobiotiques. Les métabolites formés dans un organe peuvent être ensuite métabolisés à nouveau dans un second organe. Les bactéries intestinales peuvent aussi participer à la biotransformation.
Les métabolites des xénobiotiques peuvent être excrétés par les reins ou par la bile. Ils peuvent aussi être exhalés par les poumons, ou se lier à des molécules endogènes dans l’organisme.
La relation entre la biotransformation et la toxicité est complexe. La biotransformation peut être considérée comme un processus nécessaire à la survie. Elle protège l’organisme vis-à-vis d’une toxicité en empêchant les substances nocives de s’accumuler dans l’organisme. Cependant, des métabolites réactifs intermédiaires peuvent se former lors de la biotransformation, métabolites qui sont potentiellement dangereux. Ce phénomène est appelé l’activation métabolique. La biotransformation peut donc induire également une toxicité. S’ils ne sont pas conjugués, les métabolites oxydés intermédiaires peuvent se lier aux structures cellulaires et les endommager. Par exemple, la liaison d’un métabolite de xénobiotique à l’ADN peut être à l’origine d’une mutation (voir l’article «La toxicologie génétique»). Si le système de biotransformation est dépassé, il peut se produire une destruction massive de protéines essentielles ou des membranes lipidiques qui peut aboutir à la mort cellulaire (voir l’article «La lésion et la mort cellulaires»).
Le terme métabolisme est souvent employé de façon interchangeable avec celui de biotransformation. Il désigne la dégradation chimique ou les réactions de synthèse catalysées par des enzymes dans l’organisme. Les nutriments alimentaires, les composés endogènes et les xénobiotiques sont tous métabolisés dans l’organisme.
L’activation métabolique signifie qu’un composé moins réactif est converti en une molécule plus réactive. Cette conversion se produit lors des réactions de phase I.
L’inactivation métabolique renvoie au fait qu’une molécule active ou toxique est convertie en un métabolite moins actif. Ce phénomène se produit généralement lors des réactions de phase II. Dans certains cas, un métabolite inactivé peut être réactivé, par suite d’un clivage enzymatique, par exemple.
La réaction de phase I, qui constitue la première étape du métabolisme d’un xénobiotique, indique généralement que le composé est oxydé. L’oxydation crée habituellement un composé plus hydrosoluble et facilite les réactions ultérieures.
Les enzymes du cytochrome P450 constituent un groupe d’enzymes oxydant préférentiellement les xénobiotiques lors des réactions de phase I. Les différentes enzymes sont spécialisées pour la prise en charge de groupes spécifiques de xénobiotiques présentant certaines caractéristiques. Les molécules endogènes sont également des substrats pour ces enzymes. Les enzymes du cytochrome P450 sont induites par des xénobiotiques d’une manière spécifique. La connaissance d’une induction du cytochrome P450 peut renseigner utilement sur la nature des expositions antérieures (voir l’article «Les déterminants génétiques de la réponse toxique»).
La réaction de phase II, qui représente la seconde étape dans le métabolisme des xénobiotiques, signifie que le composé oxydé est conjugué (couplé) à une molécule endogène. Cette réaction se caractérise par une augmentation de l’hydrosolubilité. De nombreux métabolites conjugués sont fortement excrétés par la voie rénale.
Les transférases constituent un groupe d’enzymes catalysant les réactions de phase II. Elles conjuguent les xénobiotiques avec des composés endogènes tels que le glutathion, les acides aminés, l’acide glucuronique ou le sulfate.
Le glutathion est une molécule endogène, un tripeptide, conjugué aux xénobiotiques lors des réactions de phase II. Il est présent dans toutes les cellules (en fortes concentrations dans les cellules hépatiques) et, généralement, protège de la toxicité des xénobiotiques activés. Lorsque le glutathion est épuisé, des réactions toxiques peuvent se produire entre les métabolites actifs des xénobiotiques et les protéines, les lipides ou l’ADN.
L’induction signifie que les enzymes participant à la biotransformation sont augmentées (en activité ou en quantité) en réponse à l’exposition à un xénobiotique. Dans certains cas, l’activité peut subir plusieurs augmentations en quelques jours. L’induction est souvent équilibrée lorsque les réactions des phases I et II subissent simultanément une augmentation; il se produira alors une biotransformation plus rapide qui peut expliquer une tolérance. Au contraire, une induction déséquilibrée peut accroître la toxicité.
L’inhibition de la biotransformation peut survenir lorsque deux xénobiotiques sont métabolisés par la même enzyme. Les deux substrats entrent en compétition et, généralement, l’un des substrats l’emporte. Dans ce cas, le second substrat n’est pas métabolisé, ou l’est plus lentement. Comme pour l’induction, l’inhibition peut donc faire augmenter la toxicité ou la faire diminuer.
L’activation de l’oxygène peut être déclenchée par les métabolites de certains xénobiotiques. Ils peuvent s’auto-oxyder en produisant des espèces oxygénées activées. Ces espèces dérivées de l’oxygène, qui incluent le superoxyde, le peroxyde d’hydrogène et le radical hydroxyle, peuvent léser l’ADN, les lipides et les protéines dans les cellules. L’activation de l’oxygène intervient également dans les processus inflammatoires.
La variabilité génétique entre les individus a été constatée pour de nombreux gènes codant pour des enzymes de phase I et de phase II. Cette variabilité peut expliquer que certains individus soient plus sensibles que d’autres aux effets toxiques des xénobiotiques.
L’organisme humain constitue un système biologique complexe avec des niveaux d’organisation variés, depuis le niveau moléculaire-cellulaire jusqu’aux tissus et organes. Il s’agit d’un système ouvert, échangeant matière et énergie avec l’environnement à travers des réactions biochimiques nombreuses en équilibre dynamique, environnement qui peut être pollué ou contaminé par divers toxiques.
La pénétration de molécules ou d’ions toxiques depuis l’environnement, général ou professionnel, dans un tel système biologique si fortement coordonné, peut perturber de manière réversible ou irréversible les processus biochimiques cellulaires normaux, ou même léser et détruire la cellule (voir l’article «La lésion et la mort cellulaires»).
La pénétration d’un toxique depuis l’environnement jusqu’aux sites où il va exercer son effet toxique dans l’organisme peut être divisé en trois phases:
Nous nous intéresserons plus particulièrement aux processus toxicocinétiques qui ont lieu dans l’organisme humain après une exposition à des toxiques environnementaux.
Les molécules ou les ions toxiques présents dans l’environnement pénètrent dans l’organisme à travers la peau et les muqueuses, ou les cellules épithéliales des appareils respiratoire et gastro-intestinal, selon la voie d’entrée. Pour cela, les molécules et les ions toxiques doivent franchir les membranes cellulaires de ces systèmes biologiques ainsi que le réseau complexe des membranes se trouvant à l’intérieur de la cellule.
Tous les processus toxicocinétiques et toxicodynamiques se produisent au niveau moléculaire ou cellulaire. Ils sont commandés par un certain nombre de facteurs que l’on peut scinder en deux groupes fondamentaux:
C’est en 1854 que le toxicologue russe E.V. Pelikan a commencé l’étude de la relation entre la structure chimique d’une substance et son activité biologique — la relation structure-activité (RSA). La structure chimique détermine directement les propriétés physico-chimiques, dont certaines sont responsables de l’activité biologique.
Pour définir la structure chimique, on a le choix entre plusieurs paramètres ou descripteurs qui peuvent être répartis selon les groupes suivants:
Pour chaque toxique, il convient donc de sélectionner un ensemble de descripteurs correspondant à un mécanisme d’activité particulier. Néanmoins, du point de vue toxicocinétique, deux paramètres revêtent une importance générale pour tous les toxiques:
Dans le cas des aérosols et des poussières inhalées, leur toxicocinétique et leur toxicodynamique est aussi fonction de la taille des particules, de leur forme, de leur surface et de leur densité.
La cellule eucaryote est entourée d’une membrane cytoplasmique qui commande le transport des substances et maintient l’homéostasie cellulaire. Les organites cellulaires (noyau, mitochondrie) possèdent eux aussi une membrane. Le cytoplasme cellulaire est compartimenté par des structures membranaires intriquées, le réticulum endoplasmique et l’appareil de Golgi (endomembranes). Toutes ces membranes ont une structure semblable, mais diffèrent par leur teneur en lipides et en protéines.
Structurellement, les membranes sont constituées d’une double couche de molécules lipidiques (phospholipides, sphingolipides, cholestérol). La principale composante de la molécule de phospholipide est le glycérol dont deux groupes OH sont estérifiés par des acides gras aliphatiques de 16 à 18 atomes de carbone, le troisième groupe étant estérifié par un groupe phosphate et un composé azoté (choline, éthanolamine, sérine). Les sphingolipides quant à eux sont surtout formés de sphingosine.
La molécule lipidique est amphipathique, car elle possède une «tête» polaire hydrophile (amino-alcool, phosphate, glycérol) et une double «queue» non polaire (acides gras). La double couche lipidique est disposée de telle sorte que les têtes hydrophiles constituent la surface intérieure et extérieure de la membrane et que les queues lipophiles sont étirées vers l’intérieur de la membrane, qui contient de l’eau, divers ions et des molécules.
Des protéines et des glycoprotéines sont insérées dans la double couche lipidique (protéines intrinsèques) ou attachées à la surface de la membrane (protéines extrinsèques). Ces protéines contribuent à l’intégrité structurale de la membrane, mais elles peuvent également remplir la fonction d’enzymes, de protéines porteuses, de parois de pores ou de récepteurs.
La membrane constitue une structure dynamique qui, selon les besoins fonctionnels, peut être désagrégée et reconstruite avec une proportion différente de lipides et de protéines.
Le contrôle du transport des substances à l’intérieur et à l’extérieur de la cellule constitue l’une des fonctions fondamentales des membranes externes et internes.
Certaines molécules lipophiles passent directement à travers la double couche lipidique, les molécules hydrophiles et les ions étant transportés par les pores. Les membranes réagissent au changement de conditions en ouvrant ou en fermant des pores de diverses tailles.
Les processus et mécanismes suivants interviennent dans le transport de substances, y compris celui des toxiques, à travers les membranes:
Processus actifs:
Elle représente le mouvement des molécules et des ions à travers la double couche lipidique ou les pores depuis une région à forte concentration, ou à fort potentiel électrique, vers une région à faible concentration ou potentiel («gradient de concentration»). La différence de concentration ou de charge électrique est la force motrice déterminant l’intensité du flux dans les deux directions. A l’état d’équilibre, l’afflux est égal au flux sortant. La diffusion est régie par la loi de Fick, selon laquelle le taux est directement proportionnel à la surface membranaire disponible, au gradient de concentration (charge) et à un coefficient de diffusion, et inversement proportionnel à l’épaisseur de la membrane.
Les petites molécules lipophiles passent facilement à travers la couche lipidique membranaire selon le coefficient de partage de Nernst.
Les grosses molécules lipophiles, les molécules hydrosolubles et les ions utilisent les pores aqueux pour leur passage. La taille et la configuration stérique conditionnent le passage des molécules. Pour les ions, outre la taille, le type de charge est déterminant. Les protéines constitutives de la paroi des pores peuvent acquérir une charge positive ou négative. Les pores étroits sont sélectifs — les ligands chargés négativement permettant le seul passage des cations, les ligands chargés positivement uniquement celui des anions. Lorsque le diamètre du pore augmente, le flux hydrodynamique est dominant et permet le libre passage des ions et des molécules, selon la loi de Poiseuille. La filtration est une conséquence du gradient osmotique. Dans certains cas, les ions peuvent pénétrer par l’intermédiaire de molécules spécifiques complexes — les ionophores — produits par des micro-organismes et présentant des effets antibiotiques (nonactine, valinomycine, gramicidine, etc.).
Ce type de diffusion requiert la présence d’une molécule porteuse dans la membrane, généralement de nature protéique (perméase). La molécule porteuse fixe les substances d’une manière sélective, ressemblant à un complexe substrat-enzyme. Des molécules similaires (y compris des toxiques) peuvent entrer en compétition vis-à-vis de la molécule porteuse spécifique jusqu’à ce que le point de saturation soit atteint. Des toxiques peuvent entrer en compétition vis-à-vis de la molécule porteuse; une fois liés à celle-ci de façon irréversible, le transport est bloqué. Chaque type de molécule porteuse présente un taux de transport caractéristique. S’il est réalisé dans les deux directions, le transport est appelé diffusion d’échange.
Dans le cas de certaines substances vitales pour la cellule, un type spécial de transporteur existe, qui permet le transport contre le gradient de concentration ou le potentiel électrique («en amont»). La molécule porteuse présente une grande stéréospécificité et elle est saturable.
Ce type de transport nécessite de l’énergie, qui lui est fournie par le clivage catalytique de molécules d’ATP en molécules d’ADP par l’enzyme adénosine triphosphatase (ATP-ase).
Des toxiques peuvent interférer avec ce type de transport par inhibition compétitive ou non compétitive des molécules porteuses ou par inhibition de l’activité ATP-asique.
L’endocytose est un mécanisme de transport au cours duquel la membrane cellulaire enveloppe le matériau pour former une vésicule pénétrant dans la cellule. Lorsque le matériau concerné est liquide, le processus est appelé pinocytose. Dans certains cas, ce matériau est lié à un récepteur et le complexe ainsi formé est transporté par une vésicule membranaire. C’est ce type de transport qu’utilisent notamment les cellules épithéliales du tractus gastro-intestinal et les cellules hépatiques et rénales.
L’organisme est exposé à de nombreux toxiques présents dans l’environnement général ou professionnel. Ces toxiques peuvent pénétrer dans l’organisme par trois portes d’entrée principales:
Dans l’industrie, l’inhalation représente la principale porte d’entrée des toxiques, suivie de la pénétration cutanée. Dans l’agriculture, l’absorption en cas d’exposition à certains pesticides se fait autant par la peau que par inhalation. Quant à la population dans son ensemble, elle est exposée par voie gastro-intestinale essentiellement (ingestion de nourriture, d’eau et de boissons contaminées), mais aussi par voie inhalatoire et, plus rarement, par pénétration cutanée.
L’absorption pulmonaire représente la principale voie de captation de nombreux toxiques présents dans l’air (gaz, vapeurs, fumées, brouillards, poussières, aérosols, etc.).
L’appareil respiratoire constitue un système idéal pour les échanges gazeux. Il présente en effet une surface membranaire totale allant de 30 m2 (à l’expiration) à 100 m2 (lors d’une inspiration profonde), faisant face à un réseau capillaire d’environ 2 000 km. Le système, développé au cours de l’évolution, est localisé dans un espace relativement petit (cavité thoracique) protégé par les côtes.
Anatomiquement et physiologiquement, l’appareil respiratoire peut être divisé en trois compartiments:
Les toxiques hydrophiles sont facilement absorbés par l’épithélium de la région nasopharyngienne, l’épithélium des régions nasopharyngienne et trachéo-bronchique étant recouvert en totalité d’un film aqueux. Les toxiques lipophiles sont un peu absorbés dans ces deux régions, mais ils le sont principalement au niveau des alvéoles par diffusion à travers les membranes alvéolo-capillaires. Le taux d’absorption dépend de la ventilation pulmonaire, du débit cardiaque (qui conditionne le flux sanguin au niveau pulmonaire), de la solubilité du toxique dans le sang et de son métabolisme.
C’est au niveau alvéolaire que s’effectuent les échanges gazeux. La paroi alvéolaire est constituée d’un épithélium, d’une membrane basale interstitielle, de tissu conjonctif et d’un endothélium capillaire. A travers ces couches dont l’épaisseur est de 0,8 µm environ, la diffusion des toxiques est très rapide. Dans les alvéoles, le toxique est échangé entre la phase aérienne et la phase liquide (sang). Le taux d’absorption d’un toxique (distribution de l’air vers le sang) dépend de sa concentration dans l’air alvéolaire et du coefficient de partage de Nernst pour le sang (coefficient de solubilité).
Dans le sang, le toxique est dissous dans la phase liquide par simple processus physique ou par suite de sa liaison aux cellules sanguines ou aux constituants plasmatiques selon l’affinité chimique ou par adsorption. Le sang contenant 75% d’eau, les gaz et les vapeurs hydrophiles présentent donc une grande solubilité dans le plasma (par exemple, les alcools). Les toxiques lipophiles (comme le benzène) sont généralement liés aux cellules ou aux macromolécules telles que l’albumine.
Dès le début d’une exposition par voie pulmonaire, deux processus opposés surviennent: l’absorption et la désorption. L’équilibre entre ces processus dépend de la concentration du toxique dans l’air alvéolaire et le sang. En début d’exposition, la concentration sanguine en toxiques est nulle et la rétention dans ce milieu est pratiquement totale. Avec la poursuite de l’exposition, un équilibre s’établit entre l’absorption et la désorption. Les toxiques hydrophiles atteignent rapidement l’équilibre et le taux d’absorption dépend de la ventilation pulmonaire plutôt que du flux sanguin. Les toxiques lipophiles ont besoin d’un temps plus long pour atteindre l’équilibre et, dans ce cas, le flux de sang insaturé commande le taux d’absorption.
Le dépôt des particules et des aérosols dans le tractus respiratoire dépend de facteurs physiques et physiologiques et de la taille des particules. Plus la particule est petite, plus elle pénètre profondément dans le tractus respiratoire.
La rétention relativement faible des particules de poussière observée de façon constante dans les poumons de personnes fortement exposées (les mineurs, par exemple) donne à penser qu’il existe un système très efficace de clairance des particules. Dans la partie supérieure du tractus respiratoire (zone trachéo-bronchique), cette fonction est assurée par une couche mucociliaire. Dans la partie pulmonaire, trois mécanismes ou niveaux interviennent: 1) la couche mucociliaire; 2) la phagocytose; 3) la pénétration directe des particules à travers la paroi alvéolaire.
Les 17 premières des 23 arborescences de l’arbre trachéo-bronchique possèdent des cellules épithéliales ciliées. Par leurs mouvements, ces cils poussent continuellement une couche de mucus vers la bouche. Les particules déposées sur cette couche mucociliaire sont avalées au niveau buccal (ingestion). Une couche de mucus couvre également la surface de l’épithélium alvéolaire, se déplaçant en direction de la couche mucociliaire. De plus, des cellules spécialisées pouvant se déplacer — les phagocytes — absorbent les particules et les micro-organismes présents dans les alvéoles et migrent dans deux directions possibles:
Les toxiques peuvent être ingérés à la suite d’une ingestion accidentelle, de l’absorption de nourriture ou de boissons contaminées, ou par ingestion de particules éliminées par le tractus respiratoire.
Le tractus digestif dans son intégralité, depuis l’œsophage jusqu’à l’anus, présente la même structure de base. Une couche muqueuse (épithélium) est sous-tendue de tissu conjonctif et, au-delà, par un réseau de capillaires et de muscle lisse. La surface de l’épithélium stomacal est très plissée ce qui accroît la surface d’absorption et de sécrétion. La surface intestinale contient de nombreux replis (villosités), capables d’absorber le matériel par «pompage». La surface active pour l’absorption dans les intestins est d’environ 100 m2.
Au niveau du tractus gastro-intestinal, tous les processus d’absorption sont très actifs:
Certains ions de métaux toxiques utilisent les systèmes de transport spécialisés des éléments essentiels: le thallium, le cobalt et le manganèse font appel au système de transport du fer, le plomb employant celui du calcium.
De nombreux facteurs ont une influence sur le taux d’absorption des toxiques dans les diverses parties du tractus gastro-intestinal:
Il faut également mentionner la circulation entéro-hépatique. Les toxiques ou leurs métabolites polaires (glucuronides et autres conjugués) sont excrétés avec la bile dans le duodénum. A ce niveau, les enzymes de la microflore réalisent une hydrolyse et les produits libérés peuvent être réabsorbés et transportés par la veine porte vers le foie. Ce mécanisme est très dangereux dans le cas de substances hépatotoxiques, car il permet leur accumulation temporaire dans le foie.
S’agissant des toxiques biotransformés dans le foie en métabolites moins toxiques ou non toxiques, l’ingestion peut représenter une voie d’entrée moins dangereuse. Après absorption dans le tractus gastro-intestinal, ces toxiques sont transportés par la veine porte au foie où ils peuvent être partiellement détoxifiés par biotransformation.
La peau (1,8 m2 de surface chez l’adulte) et les muqueuses des orifices recouvrent la surface corporelle. La peau agit comme un rempart vis-à-vis des agents physiques, chimiques et biologiques et, entre autres tâches physiologiques, elle maintient l’intégrité du corps et l’homéostasie.
La peau est constituée de trois couches: l’épiderme, la vraie peau (le derme) et les tissus sous-cutanés (hypoderme). Du point de vue toxicologique, l’épiderme est du plus grand intérêt. Il est constitué de nombreuses couches cellulaires. Une surface calleuse de cellules mortes aplaties (stratum corneum, couche cornée) constitue la couche supérieure, sous laquelle se trouvent une couche continue de cellules vivantes (stratum corneum compactum, couche cornée compacte), une membrane lipidique typique, puis le stratum lucidum, le stratum granulosum et le stratum mucosum. La membrane lipidique représente une barrière protectrice que traversent les follicules des poils et les canaux des glandes sudoripares dans les parties velues de la peau. L’absorption dermique peut donc se faire selon les mécanismes suivants:
Le taux d’absorption à travers la peau dépend de nombreux facteurs:
Quelle que soit la voie d’absorption, les toxiques atteignent le sang, la lymphe ou les autres fluides corporels. Le sang représente le véhicule principal assurant le transport des toxiques et de leurs métabolites.
Le sang, tissu liquide fluide circulant, transporte l’oxygène et les nutriments nécessaires aux cellules et élimine les produits de déchet du métabolisme. Il contient également des composants cellulaires, des hormones et d’autres molécules participant à de nombreuses fonctions physiologiques. Le sang circule à l’intérieur d’un réseau de vaisseaux sanguins, relativement bien fermé et sous pression élevée en raison de l’activité cardiaque. Cette pression élevée entraîne une fuite liquidienne et le système lymphatique fait office de système de drainage grâce à un fin réseau de petits capillaires lymphatiques à paroi mince ayant des ramifications dans les tissus mous et les organes.
Le sang est le mélange d’une phase liquide (plasma, 55%) et d’une phase solide constitué de cellules sanguines (45%). Le plasma contient des protéines (albumines, globulines, fibrinogène), des acides organiques (lactique, glutamique, citrique) et de nombreuses autres substances (lipides, lipoprotéines, glycoprotéines, enzymes, sels, xénobiotiques, etc.). Les cellules sanguines comprennent les érythrocytes, les leucocytes, les réticulocytes, les monocytes et les plaquettes.
Les toxiques sont absorbés sous forme moléculaire ou ionique. Certains d’entre eux forment, au pH du sang, des particules colloïdales constituant la troisième forme de transport dans ce liquide. Les molécules, les ions et les colloïdes toxiques sont transportés dans le sang de diverses manières:
La plupart des toxiques sanguins se trouvent soit à l’état libre dans le plasma, soit liés aux érythrocytes et aux constituants plasmatiques. Leur distribution dépend de leur affinité envers ces constituants. Toutes les fractions sont en équilibre dynamique.
Certains toxiques sont transportés par les éléments du sang — la plupart par les érythrocytes — très rarement par les leucocytes. Les toxiques peuvent être adsorbés à la surface des érythrocytes ou se lier aux ligands du stroma. S’ils pénètrent dans les érythrocytes, ils peuvent se lier à l’hème (le monoxyde de carbone et le sélénium, par exemple) ou à la globine (Sb111, Po210). Parmi les toxiques transportés par les érythrocytes, on trouve l’arsenic, le césium, le plomb, le radium, le sodium et le thorium. Le chrome hexavalent est exclusivement lié aux érythrocytes et le chrome trivalent aux protéines plasmatiques. Dans le cas du zinc, on assiste à une concurrence entre les érythrocytes et le plasma. Le plomb est transporté à 96% environ par les érythrocytes. Le mercure organique est principalement lié aux érythrocytes, le mercure inorganique étant en majeure partie acheminé par l’albumine plasmatique. De petites fractions de béryllium, de cuivre, de tellure et d’uranium sont prises en charge par les érythrocytes.
La majorité des toxiques sont transportés par le plasma ou les protéines plasmatiques. De nombreux électrolytes sont présents sous forme ionique en équilibre avec les molécules non ionisées libres ou associées aux fractions plasmatiques. La fraction ionique des toxiques est très diffusible et pénètre à travers les parois des capillaires dans les tissus et organes. Les gaz et vapeurs peuvent être dissous dans le plasma.
Les protéines plasmatiques possèdent une surface totale d’environ 600 à 800 km2 pouvant assurer l’absorption des toxiques. Les molécules d’albumine possèdent environ 109 ligands cationiques et 120 ligands anioniques à la disposition des ions. De nombreux ions sont partiellement transportés par l’albumine (cadmium, cuivre et zinc, par exemple). Il en va de même pour les composés tels que les dinitro- et ortho-crésols, les dérivés nitrés et halogénés des hydrocarbures aromatiques et les phénols.
Les molécules de globuline (alpha et bêta) transportent des toxiques de faible poids moléculaire, des ions métalliques (cuivre, fer et zinc) et des particules colloïdales. Le fibrinogène a une affinité pour les molécules de faible poids moléculaire. Divers types de liaisons peuvent se former entre les toxiques et les protéines plasmatiques: forces de van der Waals, attraction de charges, association entre groupes polaires et apolaires, ponts hydrogène, liaisons covalentes.
Les lipoprotéines plasmatiques transportent des toxiques lipophiles comme les PCB. Les autres fractions plasmatiques interviennent aussi dans ce transport. L’affinité des toxiques pour les protéines plasmatiques témoigne de leur affinité protéique dans les tissus et les organes lors de la distribution.
Les acides organiques (lactique, glutamique, citrique) forment des complexes avec certains toxiques. Les éléments alcalins et les terres rares, de même que certains éléments lourds sous forme cationique, sont également complexés avec des oxyacides organiques et des acides aminés. Tous ces complexes sont généralement diffusibles et facilement distribués dans les tissus et les organes.
Physiologiquement, les agents chélateurs plasmatiques tels que la transferrine et la métallothionéine rivalisent avec les acides organiques et les acides aminés vis-à-vis des cations pour former des chélates stables.
Les ions libres diffusibles et certains complexes et molécules libres passent facilement du sang aux tissus et aux organes. La fraction libre des ions et des molécules est en équilibre dynamique avec la fraction liée. La distribution d’un toxique du sang vers les tissus et les organes ou, inversement, sa mobilisation depuis les tissus et les organes vers le sang, dépendent de sa concentration sanguine.
L’organisme humain peut être divisé en plusieurs compartiments: 1) les organes internes; 2) la peau et les muscles; 3) le tissu adipeux; 4) le tissu conjonctif et le tissu osseux. Cette classification est principalement basée sur le degré, en l’occurrence décroissant, d’irrigation vasculaire (sanguine). Ainsi, les organes internes (dont le cerveau), représentant 12% du poids corporel total, reçoivent environ 75% du volume sanguin total. A l’opposé, les tissus conjonctif et osseux (15% du poids total du corps) ne reçoivent que 1% du volume sanguin total.
Les organes internes fortement irrigués atteignent généralement la plus forte concentration toxique dans le temps le plus court; de même, l’état d’équilibre entre ces organes et le sang est atteint plus rapidement. La captation des toxiques par les tissus moins perfusés est plus lente, mais la rétention y est plus forte et la durée de séjour plus longue (accumulation) en raison de la faible perfusion.
Trois éléments revêtent une importance capitale dans la distribution intracellulaire des toxiques: l’eau, les lipides et les protéines, et en particulier leur teneur dans les cellules des divers tissus et organes. Les compartiments susmentionnés se caractérisent par une teneur en eau cellulaire décroissante. Les toxiques hydrophiles sont distribués plus rapidement dans les fluides et les cellules riches en eau, alors que la distribution des toxiques lipophiles est plus rapide vers les cellules à contenu lipidique élevé (tissus gras).
L’organisme possède des barrières empêchant la pénétration de certains groupes de toxiques, surtout hydrophiles, dans des organes et des tissus:
Comme nous l’avons déjà mentionné, seules les formes libres des toxiques dans le plasma (molécules, ions, colloïdes) peuvent pénétrer à travers les parois capillaires. Cette fraction libre est en équilibre dynamique avec la fraction liée. La concentration des toxiques dans le sang, qui est elle aussi en équilibre dynamique avec leur concentration dans les organes et les tissus, commande leur rétention (accumulation) ou leur mobilisation dans ces milieux.
L’état général de l’organisme, l’état fonctionnel des organes (en particulier la régulation neuro-humorale), l’équilibre hormonal et d’autres facteurs jouent un rôle dans la distribution.
La rétention d’un toxique dans un compartiment donné est généralement temporaire et se termine par une redistribution vers d’autres tissus. La rétention et l’accumulation sont basées sur les différences entre vitesse d’absorption et vitesse d’élimination. La durée de rétention dans un compartiment est exprimée par la demi-vie biologique, intervalle de temps durant lequel 50% du toxique sont éliminés du tissu ou de l’organe pour être redistribués dans l’organisme ou en être éliminés.
Lors de la distribution et de la rétention dans les organes et tissus, on asssiste à divers processus de biotransformation. Cette biotransformation produit des métabolites plus polaires et plus hydrophiles, qui sont plus faciles à éliminer. Un taux faible de biotransformation d’un toxique lipophile provoque généralement son accumulation dans un compartiment.
Les toxiques peuvent être divisés en quatre groupes principaux selon leur affinité et leur mode prédominant de rétention et d’accumulation dans un compartiment particulier:
Un homme «normal» de 70 kg de poids corporel est constitué de 15% environ de tissu adipeux (jusqu’à 50% chez l’obèse), mais cette fraction lipidique n’est pas répartie uniformément. Le cerveau (SNC) est un organe riche en lipides et les nerfs périphériques sont entourés d’une gaine de myéline riche en lipides et en cellules de Schwann, tissus qui tous permettent l’accumulation de toxiques lipophiles.
De nombreux toxiques non ionisés et apolaires ayant un coefficient de partage de Nernst favorable seront distribués dans ce compartiment, de même que de nombreux solvants organiques (alcools, aldéhydes, cétones, etc.), des hydrocarbures chlorés (dont les insecticides organochlorés comme le DDT), certains gaz inertes (radon), etc.
Le tissu adipeux accumule les toxiques en raison de sa vascularisation et de son taux de biotransformation faibles. L’accumulation des toxiques peut y représenter une sorte de «neutralisation» temporaire du fait de l’absence de cibles pour l’effet toxique dans ce milieu. Cependant, le danger potentiel pour l’organisme est toujours présent en raison de la possibilité d’une mobilisation des toxiques depuis ce compartiment vers la circulation.
Le dépôt de toxiques au niveau cérébral (SNC) ou dans le tissu riche en lipides de la gaine de myéline du système nerveux périphérique s’avère très nocif. En effet, les neurotoxiques sont déposés directement à proximité de leur cible. Les toxiques retenus dans les tissus riches en lipides des glandes endocrines peuvent entraîner des troubles hormonaux. Malgré la barrière hémato-encéphalique, de nombreux neurotoxiques lipophiles atteignent le cerveau (SNC): anesthésiques, organomercuriels, pesticides, plomb tétraéthyle, solvants organiques, etc.
Dans tous les tissus et organes, des cellules spéciales possèdent une activité phagocytaire leur permettant de piéger les micro-organismes, les particules, les particules colloïdales, etc. Ce système, appelé système réticulo-endothélial, comporte à la fois des cellules fixes et des cellules mobiles (phagocytes) présentes sous forme inactive. Lorsqu’elles se trouvent exposées à un nombre élevé de microbes ou de particules, ces cellules sont activées jusqu’à un point de saturation.
Les toxiques colloïdaux sont captés par le système réticulo-endothélial des organes et des tissus. La distribution dépend de la taille des particules colloïdales, la rétention des plus grosses particules ayant lieu préférentiellement dans le foie. Pour les particules colloïdales plus petites, une distribution plus ou moins uniforme se fait entre la rate, la moelle osseuse et le foie. Au niveau du SNC, la clairance des colloïdes est très lente, alors que les petites particules sont éliminées de façon relativement plus rapide.
Environ 60 éléments sont identifiés comme éléments ostéotrophiques, ou «chercheurs d’os».
Les éléments ostéotrophiques peuvent être divisés en trois groupes:
Le squelette d’un homme normal représente 10 à 15% du poids corporel total et constitue un potentiel de stockage important pour les toxiques ostéotrophiques. L’os est un tissu hautement spécialisé formé, en volume, de 54% de minéraux et de 38% de matrice organique. La matrice minérale osseuse est constituée d’hydroxyapatite, Ca10(PO4)6(OH)2, dans laquelle le rapport Ca/P est d’environ 1,5 à 1. La surface de minéral disponible pour l’adsorption est d’environ 100 m2 par gramme de tissu osseux.
Les os du squelette peuvent être divisés en deux catégories en fonction de leur activité métabolique:
Chez le fœtus, le nouveau-né et le jeune enfant, l’os métabolique («squelette disponible») représente près de 100% du squelette. Ce pourcentage d’os métabolique décroît avec l’âge. L’incorporation des toxiques lors d’une exposition se fait dans l’os métabolique et dans les compartiments se renouvelant plus lentement.
Cette incorporation se produit de deux manières:
L’os minéral, hydroxyapatite, représente un système complexe d’échange ionique. Les ions calcium peuvent être échangés avec divers cations. Les anions présents dans l’os peuvent également être échangés par des anions: le phosphate par des citrates et des carbonates, l’hydroxyle par des fluorures. Les ions non échangeables peuvent être adsorbés à la surface minérale. Lorsque des ions toxiques sont incorporés dans le minéral, une nouvelle couche de minéral peut recouvrir la précédente, emprisonnant le toxique dans la structure osseuse. L’échange ionique est un processus réversible, dépendant de la concentration en ions, du pH et du volume de fluide. Ainsi, une augmentation en calcium alimentaire peut faire diminuer le dépôt d’ions toxiques dans le réseau minéral. Avec l’âge, le pourcentage d’os métabolique baisse, alors que l’échange ionique se poursuit; on assiste alors à une résorption osseuse, au cours de laquelle la densité osseuse décroît. Les toxiques présents dans l’os peuvent alors être relargués (plomb, par exemple).
Environ 30% des ions incorporés dans les os sont faiblement fixés et peuvent être échangés, capturés par des agents chélateurs naturels, puis excrétés, avec une demi-vie biologique de 15 jours. Les 70% restants sont fixés plus solidement, leur mobilisation et leur excrétion ayant une demi-vie biologique de 2,5 années ou plus selon le type d’os (processus de remodelage).
Les agents chélateurs (Ca-EDTA, pénicillamine, BAL, etc.) peuvent mobiliser des quantités très importantes de métaux lourds et entraîner une forte augmentation de leur excrétion urinaire.
Les particules colloïdales sont adsorbées à la manière d’un film sur la surface minérale (100 m2 par g) par des forces de van der Waals ou par adsorption chimique. Cette couche colloïdale est ensuite recouverte par la couche suivante de minéral et les toxiques sont alors intériorisés dans la structure osseuse. Le taux de mobilisation et d’élimination dépend des processus de remodelage.
Les cheveux et les ongles, riches en groupes thiols, contiennent de la kératine, capables de chélater les cations métalliques comme le mercure et le plomb.
La distribution de certains toxiques, en particulier les métaux lourds, à l’intérieur des cellules dans les tissus et les organes a fait l’objet d’études récentes. Grâce aux techniques d’ultracentrifugation, il est possible de séparer les diverses fractions cellulaires pour étudier leur teneur en ions métalliques et autres toxiques.
Les études chez l’animal ont montré qu’après pénétration dans la cellule, certains ions métalliques sont liés à une molécule spécifique, la métallothionéine. Cette protéine de faible poids moléculaire est présente dans les cellules hépatiques, rénales, et dans d’autres organes et tissus. Les groupes thiols de cette molécule peuvent fixer six ions par molécule. La biosynthèse de cette protéine résulte de la présence accrue d’ions métalliques. Les ions cadmium sont les plus puissants inducteurs. La métallothionéine a également pour fonction de maintenir l’homéostasie des ions vitaux comme le cuivre et le zinc. Elle peut fixer le bismuth, le cadmium, le cobalt, le cuivre, le mercure, l’or, le zinc et d’autres cations.
Durant leur rétention dans les cellules des divers tissus et organes, les toxiques sont exposés à des enzymes qui peuvent les biotransformer (métaboliser) et les transformer en métabolites. L’élimination des toxiques ou de leurs métabolites peut se faire de multiples façons: par l’air exhalé, l’urine, la bile, la sueur, la salive, le lait, les cheveux et les ongles.
Elle dépend de la voie d’entrée. Au niveau pulmonaire, le processus d’absorption/désorption débute immédiatement, les toxiques étant partiellement éliminés par l’air exhalé. Plus longue, l’élimination des toxiques absorbés par les autres voies commence après le transport sanguin et n’est complète qu’après distribution et biotransformation. Pendant l’absorption, on assiste à un équilibre entre les concentrations du toxique dans le sang et dans les tissus et organes. L’excrétion fait diminuer la concentration sanguine du toxique et peut induire sa mobilisation depuis les tissus vers le sang.
De nombreux facteurs exercent une influence sur le taux d’élimination des toxiques et de leurs métabolites:
Il est possible de distinguer deux groupes de compartiments: 1) le système d’échange rapide dans lequel la concentration tissulaire du toxique est semblable à celle du sang; 2) le système d’échange lent, où cette concentration est plus élevée que dans le sang en raison d’une liaison et d’une accumulation, au niveau du tissu adipeux, du squelette et des reins, ce qui se traduit par une rétention temporaire de certains toxiques, l’arsenic et le zinc, par exemple.
Un toxique peut être éliminé simultanément par deux ou plusieurs voies d’excrétion. Cependant, une voie est généralement prédominante.
Les scientifiques ont mis au point des modèles mathématiques décrivant l’excrétion d’un toxique donné. Ces modèles sont basés sur la mobilisation à partir d’un ou de deux compartiments (systèmes d’échange), sur la biotransformation, etc.
L’élimination par les poumons (désorption) est caractéristique des toxiques ayant une forte volatilité (solvants organiques, par exemple). Les gaz et les vapeurs de faible solubilité dans le sang seront facilement éliminés par cette voie, les toxiques de forte solubilité dans le sang l’étant par d’autres voies.
Les solvants organiques absorbés par le tractus gastro-intestinal ou la peau sont partiellement éliminés par l’air inhalé à chaque passage du sang à travers les poumons, s’ils ont une pression de vapeur suffisante. L’Alcootest utilisé pour déceler et évaluer l’alcoolémie des conducteurs fait appel à cette propriété. La concentration en oxyde de carbone dans l’air exhalé est en équilibre avec la teneur du sang en carboxyhémoglobine. Le radon, gaz radioactif, apparaît dans l’air exhalé à la suite de la baisse du radium accumulé dans le squelette.
L’élimination d’un toxique par l’air exhalé en fonction de la période postérieure à l’exposition est généralement exprimée par une courbe triphasique. La première phase correspond à l’élimination du toxique du sang, à demi-vie courte. La seconde phase, plus lente, représente l’élimination par échange sanguin avec les tissus et les organes (système d’échange rapide). La troisième phase, très lente, est due à l’échange du sang avec les tissus adipeux et le squelette. Si le toxique n’est pas accumulé dans ces derniers compartiments, la courbe sera biphasique. Dans certains cas, on peut aussi obtenir une courbe à quatre phases.
Pour établir l’exposition des travailleurs, on procède parfois au dosage des gaz et des vapeurs dans l’air exhalé au cours de la période suivant l’exposition.
Le rein est un organe spécialisé dans l’excrétion de nombreux toxiques et métabolites hydrosolubles, permettant de maintenir l’homéostasie de l’organisme. Chaque rein possède environ un million de néphrons capables d’assurer cette excrétion. L’excrétion rénale est un mécanisme très complexe comprenant trois processus:
L’excrétion d’un toxique par la voie urinaire dépend du coefficient de partage de Nernst, de la constante de dissociation et du pH urinaire, de la taille et de la forme de la molécule, de sa transformation en métabolites plus hydrophiles et de l’état de la fonction rénale.
La cinétique de l’excrétion rénale d’un toxique et de ses métabolites peut être schématisée par une courbe à deux, trois ou quatre phases, selon la distribution du toxique dans les divers compartiments corporels en fonction de la vitesse d’échange avec le sang.
Certains médicaments et ions métalliques peuvent être excrétés par la salive, par exemple, le plomb («liséré de Burton»), le mercure, l’arsenic, le cuivre, de même que les bromures et les iodures, l’alcool éthylique, les alcaloïdes, etc. Les toxiques sont ensuite ingérés pour atteindre le tractus gastro-intestinal, où ils peuvent être réabsorbés ou éliminés par les fèces.
De nombreux produits non ionisés peuvent être partiellement éliminés par la sueur: alcool éthylique, acétone, phénols, sulfure de carbone et hydrocarbures chlorés.
De nombreux métaux, solvants organiques et certains pesticides organochlorés (DDT) sont excrétés dans le lait maternel. Cette excrétion lactée peut représenter un danger pour les enfants lors de l’allaitement.
L’analyse des cheveux peut être utilisée comme indicateur de l’homéostasie pour diverses substances physiologiques. On peut évaluer par ce moyen l’exposition à certains toxiques, les métaux lourds en particulier.
L’élimination des toxiques peut être accélérée par:
Le dosage des toxiques et des métabolites dans le sang, l’air exhalé, l’urine, la sueur, les fèces et les cheveux est de plus en plus utilisé pour évaluer l’exposition humaine (tests d’exposition) ou le degré d’intoxication. C’est ainsi que des limites biologiques d’exposition (concentrations maximales admissibles (MAC)) et des indices biologiques d’exposition (Biological Exposure Indices (BEI)) ont été établis récemment. Ces dosages biologiques permettent d’évaluer l’«exposition interne» de l’organisme, c’est-à-dire l’exposition totale du corps à la fois dans l’environnement général et dans le milieu de travail, quelles que soient les voies de pénétration (voir l’article «Les indicateurs biologiques»).
Les individus sont exposés d’ordinaire, sur leur lieu de travail comme dans l’environnement général, à plusieurs agents physiques et chimiques de façon simultanée ou consécutive. En outre, certaines personnes prennent des médicaments, fument, consomment de l’alcool et de la nourriture contenant des additifs, etc., autant de facteurs à l’origine eux aussi d’expositions multiples. Les agents physiques et chimiques peuvent interagir à chaque étape des processus toxicocinétiques ou toxicodynamiques, entraînant trois effets possibles:
Les études concernant les effets combinés sont rares. Elles sont en effet très complexes en raison des nombreux facteurs et agents à prendre en compte.
En cas d’exposition à deux ou à plusieurs toxiques de manière simultanée ou consécutive, il convient donc d’envisager la possibilité que certains effets puissent se conjuguer et entraîner soit une augmentation, soit une diminution des processus toxicocinétiques.
La toxicologie professionnelle et environnementale a pour principal objectif d’améliorer la prévention des risques dus à l’exposition à des agents nocifs, dans l’environnement général ou professionnel, ou de limiter de façon notoire leurs effets sur la santé. Des systèmes ont été mis au point qui permettent d’évaluer de manière quantitative le risque lié à une exposition donnée (voir l’article «La toxicologie et les réglementations en matière de sécurité et de santé»).
Les effets d’un produit chimique sur un système ou organe donné sont liés à l’intensité et au type d’exposition: aiguë ou chronique. Etant donné la diversité des effets toxiques pouvant survenir dans un système ou organe, on a adopté vis-à-vis des organes et des effets critiques une démarche générale qui permet d’évaluer le risque et d’établir des limites de concentration à visée sanitaire pour les substances toxiques présentes dans l’environnement.
Du point de vue de la médecine préventive, il est très important d’identifier les effets nocifs précoces, pour pouvoir prévenir ou limiter l’apparition de conséquences pour la santé plus graves encore.
C’est l’approche qui a été choisie pour les métaux lourds. Ces métaux, comme le cadmium, le mercure et le plomb, appartiennent à un groupe particulier de substances dont la toxicité chronique s’exerce par accumulation dans les organes. Un groupe de travail spécialisé dans la toxicité des métaux (Nordberg, 1976) a adopté à cet égard les définitions ci-dessous.
La définition de l’organe critique proposée par ce groupe de travail a été adoptée avec une légère modification, le terme métal ayant été remplacé par l’expression substance potentiellement toxique (Duffus, 1993).
Lorsqu’on décide de considérer qu’un organe ou un système donné est critique, on tient compte non seulement du mécanisme toxique du produit dangereux, mais également de la voie d’absorption et de la population exposée.
Du point de vue biologique, on ne connaît pas toujours la signification de l’effet sous-critique; il peut servir d’indice biologique d’exposition, d’indice d’adaptation ou d’effet critique précurseur (voir l’article «Les indicateurs biologiques»). Cette dernière possibilité peut avoir un intérêt prophylactique particulier.
Le tableau 33.1 donne des exemples d’organes et d’effets critiques pour différents produits chimiques. Lors de l’exposition chronique au cadmium d’origine environnementale, où la voie d’absorption est d’une importance secondaire (les concentrations de cadmium dans l’air vont de 10 à 20 µg/m3 en zone urbaine et de 1 à 2 µg/m3 en zone rurale), l’organe critique est le rein. Dans une entreprise où la valeur limite d’exposition est de 50 µg/m3, l’inhalation représente la principale voie d’exposition, les deux organes considérés comme critiques étant le poumon et le rein.
|
Substance |
Organe critique lors d�une exposition chronique |
Effet critique |
|
Cadmium |
Poumons |
Sans seuil: |
|
Reins |
Seuil: |
|
|
|
Poumons |
Modifications fonctionnelles bénignes, emphysème |
|
Plomb |
Adultes |
Excrétion accrue d�acide δ-aminolévulinique urinaire (ALA-U); augmentation de la concentration en protoporphyrine libre érythrocytaire (PLE) |
|
|
Système nerveux périphérique |
Ralentissement des vitesses de conduction des fibres nerveuses les plus lentes |
|
Mercure (élémentaire) |
Jeunes enfants |
Diminution du QI et autres effets discrets; tremblement mercuriel (doigts, lèvres, |
|
Mercure (mercurique) |
Reins |
Protéinurie |
|
Manganèse |
Adultes |
Altération des fonctions psychomotrices |
|
Enfants |
|
|
|
|
Système nerveux central |
Altération des fonctions psychomotrices |
|
Toluène |
Muqueuses |
Irritation |
|
Chlorure de vinyle |
Foie |
Cancer (angiosarcome, risque unitaire 1 × 10�6) |
|
Acétate d�éthyle |
Muqueuses |
Irritation |
Avec le plomb, les organes critiques chez l’adulte sont le système hématopoïétique et le système nerveux périphérique, où les effets critiques (augmentation de la concentration en protoporphyrine érythrocytaire libre (PEL), augmentation de l’excrétion urinaire en acide delta-aminolévulinique, ou troubles de la conduction nerveuse périphérique) apparaissent lorsque le taux de plombémie (indice d’absorption systémique du plomb) avoisine 200 à 300µg/l. Chez le jeune enfant, où l’organe critique est le système nerveux central (SNC), des symptômes de dysfonctionnement, mis en évidence à l’aide de tests psychologiques, apparaissent dans les populations étudiées à des concentrations de l’ordre de 100 µg/l de plombémie.
D’autres définitions permettent de mieux expliciter la notion d’effet critique. Selon l’OMS (1989), l’effet critique correspond au premier effet nocif apparaissant lorsque le seuil (critique) de concentration ou de dose est atteint dans l’organe critique. Des effets nocifs comme le cancer, pour lequel aucune concentration seuil n’est définie, sont souvent considérés comme critiques. La décision de juger si un effet est critique ou non est affaire de spécialiste. Dans les lignes directrices du Programme international sur la sécurité des substances chimiques (PISSC) concernant l’établissement des documents appelés Environmental Health Criteria Documents, on définit l’effet critique comme étant l’effet nocif jugé le plus approprié pour déterminer la dose admissible. Cette définition a été formulée dans le but d’évaluer les limites d’exposition compatibles avec la santé dans l’environnement général. L’essentiel paraît donc d’établir quel effet doit être considéré comme nocif. Selon la terminologie actuelle, l’effet nocif est la modification de morphologie, physiologie, croissance, développement ou durée de vie d’un organisme résultant d’une moindre capacité à compenser un stress additionnel ou d’une augmentation de la sensibilité aux effets nocifs d’origine environnementale. Seul un expert est en mesure de juger si un effet est nocif ou non.
La figure 33.1 montre les courbes dose-réponse hypothétiques correspondant à divers effets. Lors d’une exposition au plomb, A représente un effet sous-critique (inhibition de l’ALA-déshydratase érythrocytaire), B l’effet critique (augmentation de la protoporphyrine zinc érythrocytaire ou de l’excrétion de l’acide δ-aminolévulinique), C l’effet clinique (anémie) et D l’issue fatale (décès). La dépendance existant entre les effets liés à l’exposition et la plombémie (témoin de la dose) sont évidents dans l’intoxication saturnine, aussi bien sous l’angle de la relation dose-réponse que pour les différentes variables (sexe, âge, etc.). L’établissement des effets critiques et de la relation dose-réponse pour ces effets chez l’humain permet de prévoir la fréquence d’un effet déterminé pour une dose donnée ou sa contrepartie (concentration dans le milieu biologique) dans une population donnée.
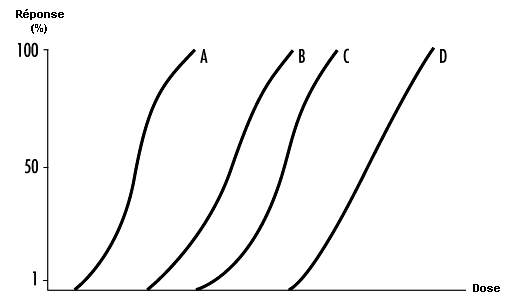
Les effets critiques peuvent être de deux types: ceux qui ont un seuil et ceux pour lesquels il existe un risque, quel que soit le niveau d’exposition (absence de seuil pour les cancérogènes génotoxiques et les mutagènes au niveau des cellules germinales). Chaque fois que possible, on doit évaluer le risque à partir de données humaines appropriées. Pour établir les seuils à appliquer à l’ensemble d’une population, des estimations du niveau d’exposition (dose admissible, indices biologiques d’exposition) doivent être faites, de sorte que la fréquence de l’effet critique dans la population exposée à un toxique donné corresponde à la fréquence de cet effet dans la population générale. Lors de l’exposition au plomb, la valeur maximale recommandée pour la plombémie dans la population générale (200 µg/l, médiane inférieure à 100 µg/l) (OMS, 1987) est pratiquement inférieure à la valeur seuil pour l’effet critique supposé — augmentation du taux de protoporphyrine érythrocytaire libre — mais cette valeur est supérieure à celle responsable des effets sur le SNC chez l’enfant ou sur la pression sanguine chez l’adulte. De façon générale, si des données obtenues à partir d’études bien conduites sur une population humaine permettent de définir un niveau pour lequel aucun effet nocif n’est observé et constituent une base d’évaluation de la sécurité, on considère qu’il convient d’appliquer un facteur d’incertitude de dix. En cas d’exposition professionnelle, les effets critiques peuvent se référer à ceux observés dans une certaine proportion de la population (par exemple, 10%). Ainsi, lors d’une exposition saturnine en milieu de travail, le niveau de plombémie admissible à visée sanitaire recommandé chez les hommes a été fixé à 400 mg/l, en partant du principe que, pour une plombémie de l’ordre de 300 à 400 mg/l, on observe une excrétion de 5 mg/l d’ALA-U chez 10% d’entre eux. S’agissant de l’exposition professionnelle au cadmium (en admettant que l’augmentation de l’excrétion urinaire de protéines de faible poids moléculaire constitue l’effet critique), on a fixé à 200 ppm la valeur admissible pour ce métal dans le cortex rénal, car cet effet critique est observé chez 10% de la population exposée. Cependant, en 1996, de nombreux pays envisageaient d’abaisser ces valeurs.
Les avis divergent quant à la méthode à suivre pour évaluer le risque de produits chimiques pour lesquels l’effet critique n’a pas de seuil, comme les cancérogènes génotoxiques. Des approches basées essentiellement sur la mise en évidence d’une relation dose-réponse ont été adoptées pour évaluer de tels effets. Du fait de l’absence de consensus sociopolitique en ce qui concerne le risque cancérogène dans des documents tels que les Directives pour la qualité de l’air en Europe (OMS, 1987), seules les valeurs du type risque unitaire rapporté à la durée de vie (c’est-à-dire le risque associé à l’exposition durant toute la vie à 1 µg/m3 de produit dangereux) sont proposées pour les effets sans seuil (voir l’article «La toxicologie et les réglementations en matière de sécurité et santé»).
Actuellement, l’étape essentielle de l’évaluation d’un risque est la détermination de l’organe et de l’effet critiques. Les définitions de l’effet critique comme de l’effet nocif témoignent du choix de l’effet à considérer comme critique dans un organe ou un système donné, choix qui est ensuite directement lié à la détermination résultante de valeurs recommandées pour un produit chimique dans l’environnement général, par exemple, dans les Directives pour la qualité de l’air en Europe (OMS, 1987) ou celle de limites à visée sanitaire applicables aux expositions professionnelles (OMS, 1980). Il peut arriver en pratique qu’il soit impossible de dire quel est l’effet critique à partir d’une gamme d’effets sous-critiques et que les limites recommandées de concentrations pour des produits chimiques toxiques dans l’environnement général ou professionnel soient impossibles à respecter. Le fait, par exemple de considérer comme critique un effet susceptible d’inclure des effets cliniques précoces peut conduire à adopter des valeurs auxquelles des effets nocifs peuvent survenir dans une partie de la population. Décider si oui ou non un effet donné doit être considéré comme critique relève de la responsabilité de groupes d’experts spécialisés en toxicologie et en évaluation du risque.
On observe souvent des différences importantes dans la manière dont les individus réagissent aux produits chimiques toxiques et parfois dans la façon dont un même individu y réagit aux différentes périodes de sa vie. Ces variations peuvent être attribuées à un grand nombre de facteurs susceptibles d’avoir une influence sur le taux d’absorption, la distribution dans l’organisme et le taux de biotransformation ou d’excrétion d’un produit chimique donné. Hormis les facteurs héréditaires qui sont connus pour avoir un lien évident avec la sensibilité aux toxiques chimiques chez l’humain (voir l’article «Les déterminants génétiques de la réponse toxique»), d’autres facteurs interviennent aussi parmi lesquels il faut citer l’âge et le sexe; un état pathologique préexistant ou la réduction fonctionnelle d’un organe (acquise); les habitudes alimentaires, le tabagisme, la consommation d’alcool et de médicaments; l’exposition concomitante à des toxines biologiques (micro-organismes) et à des facteurs physiques (rayonnements, humidité, températures très basses ou très élevées ou pressions barométriques ayant une influence particulière sur la pression partielle d’un gaz), de même que l’exercice physique ou le stress psychologique et, enfin, l’exposition préalable professionnelle ou environnementale à un produit chimique particulier, notamment l’exposition simultanée à d’autres produits chimiques non nécessairement toxiques (métaux essentiels, par exemple). La contribution possible de ces facteurs à la potentialisation ou à la diminution de la sensibilité aux effets nocifs, de même que les mécanismes d’action, varient selon le produit concerné. Par conséquent, seuls les facteurs les plus courants, les mécanismes de base et quelques exemples caractéristiques seront présentés ici, les informations spécifiques à un produit chimique particulier figurant dans une autre partie de la présente Encyclopédie.
Selon l’étape à laquelle ces facteurs interviennent (absorption, distribution, biotransformation ou excrétion d’un produit chimique particulier), on peut classer de façon schématique les mécanismes en fonction de deux types d’interaction essentiels: 1) une modification de la quantité de produit chimique au niveau de l’organe cible, c’est-à-dire au niveau de son (ses) site(s) d’effet dans l’organisme (interactions toxicocinétiques); 2) une modification de l’intensité de la réponse spécifique au produit chimique au niveau de l’organe cible (interactions toxicodynamiques). Les mécanismes les plus courants, quel que soit le type d’interaction, sont dus à la concurrence que se livrent les produits chimiques pour se lier au même constituant participant à leur transport dans l’organisme (protéines sériques spécifiques, par exemple) ou pour suivre la même voie de biotransformation (enzymes spécifiques, par exemple) aboutissant à une modification de la vitesse ou de la séquence entre la réaction initiale et l’effet nocif final. Cependant, des interactions à la fois toxicocinétiques et toxicodynamiques peuvent avoir une incidence sur la sensibilité individuelle à un produit chimique particulier. L’influence de plusieurs facteurs concomitants peut aboutir à: a) des effets additifs — l’intensité de l’effet combiné est égale à la somme des effets produits par chaque facteur séparément; b) des effets synergiques — l’intensité de l’effet combiné est supérieure à la somme des effets produits par chaque facteur séparément; c) des effets antagonistes — l’intensité de l’effet combiné est inférieure à la somme des effets produits par chaque facteur séparément.
La quantité d’un toxique ou d’un métabolite au niveau du site d’action dans l’organisme peut être évaluée approximativement par la surveillance biologique, qui consiste, par le choix du spécimen biologique adéquat et le moment optimal du prélèvement des échantillons, à prendre en compte les demi-vies biologiques d’un produit chimique donné à la fois dans l’organe critique et dans le compartiment biologique évalué. De façon générale, on manque toutefois d’informations fiables concernant les autres facteurs pouvant avoir une influence sur la sensibilité individuelle chez l’humain et, par conséquent, la majorité de nos connaissances repose sur des données expérimentales animales.
Il faut souligner qu’à niveau et durée d’exposition équivalents, l’être humain et les autres mammifères réagissent parfois de manière très différente à de nombreux toxiques; ainsi, l’humain paraît beaucoup plus sensible aux effets nocifs de plusieurs métaux toxiques que le rat (employé de façon courante dans les études expérimentales animales). Certaines de ces différences peuvent être attribuées au fait que les voies de transport, de distribution et de biotransformation de nombreux toxiques sont étroitement tributaires de variations minimes du pH tissulaire et de l’équilibre redox dans l’organisme (de la même manière que certaines activités enzymatiques). Ces disparités s’expliquent aussi par le fait que le système redox de l’être humain est très diffèrent de celui du rat.
Ces différences apparaissent clairement pour des antioxydants importants comme la vitamine C et le glutathion (GSH), essentiels au maintien de l’équilibre redox et qui, par leur rôle protecteur vis-à-vis des effets nocifs des radicaux libres dérivés de l’oxygène ou générés par des xénobiotiques, interviennent dans de nombreux états pathologiques (Kehrer, 1993). Contrairement au rat, l’être humain ne peut synthétiser la vitamine C et la teneur, de même que le taux de renouvellement du GSH érythrocytaire, sont beaucoup plus faibles chez celui-ci. Il présente aussi un déficit de certaines enzymes antioxydantes protectrices par rapport au rat et aux autres mammifères; (ainsi, la GSH-peroxydase est peu active dans le sperme humain). Ces exemples montrent bien que l’humain est beaucoup plus sensible au stress oxydatif (surtout dans les cellules sensibles, comme en témoigne la plus grande vulnérabilité du sperme humain aux toxiques, toujours par rapport au rat) et qu’il réagit de manière différente à divers facteurs auxquels il est d’ailleurs plus vulnérable que les autres mammifères (Telišman, 1995).
Comparés aux adultes, les très jeunes enfants sont souvent plus sensibles à la toxicité des substances chimiques en raison de leur volume d’inhalation relativement plus important, de leur taux d’absorption gastro-intestinale plus élevé du fait d’une plus grande perméabilité de leur épithélium intestinal, en raison aussi de l’immaturité de leurs systèmes de détoxication et d’un taux d’excrétion de produits chimiques relativement plus faible que chez l’adulte. Au début de son développement, le système nerveux central semble particulièrement sensible à la neurotoxicité de produits chimiques comme le plomb et le méthylmercure. Les sujets âgés, pour leur part, peuvent avoir été sensibilisés par des expositions antérieures responsables du stockage corporel de certains xénobiotiques ou d’une insuffisance fonctionnelle d’organes cibles ou d’activités enzymatiques, qui aboutissent à une altération des processus de détoxication et d’excrétion. Chacun de ces facteurs peut contribuer à l’affaiblissement des défenses de l’organisme et à une diminution de ses réserves, expliquant une sensibilité accrue à l’exposition ultérieure à d’autres risques. Par exemple, les enzymes du cytochrome P450 (intervenant dans la biotransformation de la plupart des produits chimiques toxiques) peuvent être induites ou inhibées sous l’influence d’un certain nombre de facteurs tout au long de la vie (dont les habitudes alimentaires, le tabagisme, l’alcool, la consommation de médicaments et l’exposition à des xénobiotiques environnementaux).
Des différences de sensibilité liées au sexe ont été décrites pour un grand nombre de produits chimiques toxiques (environ 200), différences qui se retrouvent dans de nombreuses espèces de mammifères. Les mâles sont généralement plus sensibles aux substances néphrotoxiques et les femelles aux substances hépatotoxiques. Cette différence entre mâles et femelles est non seulement liée à l’existence de disparités physiologiques (par exemple, les femelles éliminent davantage certains toxiques par voie menstruelle, par le lait maternel ou par transfert au fœtus; toutefois, elles sont soumises à un stress supplémentaire lors de la gestation, de la parturition et de la lactation), mais elle est due aussi à des disparités dans les activités enzymatiques, les mécanismes de réparation génétique, les facteurs hormonaux, ou encore à la présence chez les femelles de réserves lipidiques plus importantes, réserves responsables d’une plus grande accumulation de toxiques lipophiles, tels que les solvants organiques et certains médicaments.
Les habitudes alimentaires ont une influence importante sur la sensibilité aux toxiques, un état nutritionnel satisfaisant étant essentiel au bon fonctionnement des systèmes de défense de l’organisme vis-à-vis de ces substances. Un apport équilibré en oligoéléments (y compris les métalloïdes) et en protéines, en particulier les acides aminés soufrés, assure la biosynthèse des enzymes détoxifiantes et la fourniture de glycine et de glutathion indispensable aux réactions de conjugaison avec les composés endogènes et exogènes. Les lipides, en particulier les phospholipides, et les lipotropes (donneurs de groupements méthyles) sont nécessaires à la synthèse des membranes biologiques. Les glucides apportent l’énergie qu’exigent les processus de détoxification et fournissent l’acide glucuronique pour la conjugaison des toxiques et de leurs métabolites. Le sélénium (métalloïde essentiel), le glutathion et les vitamines telles que la vitamine C (hydrosoluble), la vitamine E et la vitamine A (liposolubles), assument le rôle important d’antioxydants (contrôle de la lipidoperoxydation et maintien de l’intégrité des membranes cellulaires) et de piégeurs de radicaux libres produits par certains toxiques. De plus, certains constituants alimentaires (protéines, fibres, minéraux, phosphates, acide citrique, etc.), de même que le volume alimentaire, peuvent avoir des répercussions non négligeables sur l’absorption gastro-intestinale de nombreux toxiques (ainsi, l’absorption moyenne des sels solubles de plomb consommés avec l’alimentation est d’environ 8%, alors qu’elle est de 60% chez le sujet à jeun). Par ailleurs, le régime alimentaire peut être également une source d’exposition individuelle supplémentaire à divers toxiques (on constate, par exemple, chez les sujets consommant des fruits de mer contaminés, une augmentation considérable des apports journaliers en arsenic, mercure, cadmium ou plomb responsables d’une accumulation).
Le tabagisme peut avoir une incidence sur la sensibilité à de nombreux toxiques en raison des interactions dues aux multiples composants présents dans la fumée de cigarette (en particulier hydrocarbures polycycliques aromatiques, monoxyde de carbone, benzène, nicotine, acroléine, certains pesticides, cadmium, et, dans une moindre mesure, plomb et autres métaux toxiques, etc.). Certains d’entre eux peuvent s’accumuler dans l’organisme tout au long de la vie, y compris durant la période prénatale (plomb et cadmium, par exemple). Les interactions ont lieu surtout au niveau du transport et de la distribution et à celui de la biotransformation à cause respectivement de la concurrence vis-à-vis des sites de liaison et vis-à-vis des enzymes qui interviennent. Ainsi, certains constituants de la fumée de cigarette peuvent induire les enzymes du cytochrome P450, alors que d’autres peuvent les inhiber, d’où une perturbation des mécanismes de biotransformation utilisés par de nombreux autres toxiques, comme les solvants organiques et certains médicaments. Une consommation importante de cigarettes sur une période prolongée peut donc affaiblir considérablement les mécanismes de défense de l’organisme en diminuant les réserves nécessaires pour faire face aux effets nocifs des autres facteurs du mode de vie.
La consommation d’alcool (éthylique) influence de diverses manières la sensibilité à de nombreux toxiques. Elle a une incidence sur l’absorption et la distribution de certains produits chimiques dans l’organisme (augmentation de l’absorption gastro-intestinale du plomb, ou encore diminution de l’absorption pulmonaire du mercure inhalé à l’état de vapeur en inhibant son oxydation nécessaire à sa rétention). L’éthanol peut également conditionner la sensibilité à divers produits chimiques en modifiant à court terme le pH tissulaire et en faisant augmenter le potentiel redox, par son métabolisme: l’oxydation de l’éthanol en acétaldéhyde, puis en acétate, produit en effet des formes réduites de nicotinamide adénine dinucléotide (NADH) et de l’hydrogène (H+). Etant donné que l’affinité de liaison tissulaire des métaux essentiels ou toxiques et des métalloïdes dépend du pH et des modifications de potentiel redox (Telišman, 1995), un apport même modéré d’éthanol peut avoir de multiples conséquences: 1) redistribution du plomb accumulé depuis longtemps dans l’organisme sous une forme inactive vers une forme active biologiquement; 2) remplacement du zinc essentiel par du plomb au niveau des enzymes contenant du zinc, modifiant ainsi l’activité enzymatique, ou influence du plomb mobilisé sur la distribution d’autres métaux et métalloïdes essentiels pour l’organisme tels que le calcium, le cuivre, le fer ou le sélénium; 3) augmentation de l’excrétion urinaire du zinc, etc. Ces effets peuvent aussi être accentués en raison de la quantité appréciable de plomb que les boissons alcoolisées peuvent renfermer à cause des contenants dans lesquels ils sont conservés ou du procédé de fabrication employé (Prpić-Majić et coll., 1984; Telišman et coll., 1984, 1993).
Autre raison courante des modifications de sensibilité liées à l’éthanol: le fait que de nombreux toxiques, des solvants organiques, par exemple, utilisent une voie de biotransformation faisant intervenir les mêmes enzymes du cytochrome P450. Selon l’intensité de l’exposition aux solvants organiques, la quantité et la fréquence de l’ingestion alcoolique (consommation aiguë ou chronique), l’éthanol peut soit ralentir, soit accélérer les vitesses de biotransformation des divers solvants organiques et modifier ainsi leur toxicité (Sato, 1991).
La consommation de médicaments peut elle aussi avoir une influence sur la sensibilité aux toxiques. Un bon nombre d’entre eux, en se liant en effet aux protéines sériques, déterminent les conditions du transport, de la distribution ou de l’excrétion de substances toxiques, et peuvent induire ou inhiber les enzymes détoxifiantes (enzymes du cytochrome P450, notamment), modifiant ainsi la toxicité des produits chimiques qui utilisent la même voie de biotransformation. L’augmentation de l’excrétion urinaire de l’acide trichloroacétique (métabolite de plusieurs hydrocarbures chlorés) après consommation de salicylés, de sulfamides ou de phénylbutazone, ou l’augmentation de l’hépato-néphrotoxicité du tétrachlorure de carbone après consommation de phénobarbital, sont caractéristiques de chacun de ces mécanismes. De plus, certains médicaments contiennent des produits chimiques potentiellement toxiques en quantité appréciable. Ainsi, les antiacides ou les préparations utilisées pour le traitement thérapeutique de l’hyperphosphatémie survenant lors d’une insuffisance rénale chronique renferment de l’aluminium.
Les modifications de la sensibilité consécutives aux interactions entre plusieurs produits chimiques (effets additifs, synergiques ou antagonistes possibles) ont surtout été étudiées chez l’animal, en particulier chez le rat. On ne dispose pas d’études épidémiologiques ou cliniques sérieuses sur la question. Cette constatation n’est pas sans conséquence quand on sait que les produits toxiques provoquent des réponses plus intenses et des effets nocifs plus divers chez l’être humain que chez le rat et les autres mammifères. La plupart des données dont on dispose, en dehors de celles publiées dans le domaine pharmacologique, concernent uniquement l’association de deux produits chimiques différents appartenant à des groupes de substances spécifiques, par exemple des pesticides, des solvants organiques, ou des métaux et métalloïdes essentiels ou toxiques.
L’exposition combinée à des solvants organiques peut provoquer des effets additifs, synergiques ou antagonistes (en fonction des associations de solvants organiques, de l’intensité et de la durée de l’exposition) dus essentiellement à leurs influences réciproques sur les processus de biotransformation (Sato, 1991).
Autre exemple caractéristique: les interactions des métaux et métalloïdes essentiels avec les agents toxiques, car elles peuvent varier en fonction de l’âge (par exemple, accumulation corporelle tout au long de la vie du plomb et du cadmium d’origine environnementale), du sexe (manque de fer habituel chez la femme), des habitudes alimentaires (apport alimentaire excessif en métaux et métalloïdes toxiques, ou apport alimentaire insuffisant en métaux et métalloïdes essentiels), des habitudes tabagiques et de la consommation d’alcool (exposition au cadmium, au plomb et à d’autres métaux toxiques, etc.), et de la consommation de médicaments (une seule dose d’antiacide pouvant, par exemple, accroître d’un facteur de 50 l’apport journalier moyen en aluminium d’origine nutritionnelle). Le tableau 33.2 illustre les diverses possibilités d’effets additifs, synergiques ou antagonistes qui peuvent survenir lors d’une exposition à des métaux et métalloïdes chez l’humain. On constate que des interactions supplémentaires peuvent se produire lorsque des éléments essentiels entrent en interaction les uns avec les autres: il faut signaler ici l’effet antagoniste bien connu du cuivre sur l’absorption gastro-intestinale et le métabolisme du zinc, et vice versa. Le mécanisme essentiel de ces interactions est la concurrence que se livrent les métaux et métalloïdes pour le même site de liaison (surtout le groupement thiol-SH) au niveau de diverses enzymes, des métalloprotéines (surtout la métallothionéine) et des tissus (membranes cellulaires et barrières entre organes). Ces interactions peuvent jouer un rôle significatif dans le développement de maladies chroniques par suite de l’action des radicaux libres et du stress oxydatif (Telišman, 1995).
|
Métal ou métalloïde toxiques |
Effets importants de l�interaction avec un autre métal ou métalloïde |
|
Aluminium (Al) |
Fait diminuer l�absorption du Ca et en modifie le métabolisme; un régime alimentaire carencé en Ca accroît le taux d�absorption de l�Al. |
|
Arsenic (As) |
Affecte la distribution du Cu (augmentation du Cu dans les reins et diminution dans le foie, le sérum et l�urine). |
|
Cadmium (Cd) |
Limite l�absorption du Ca et en modifie le métabolisme; un régime alimentaire carencé en Ca fait augmenter l�absorption du Cd. |
|
Mercure (Hg) |
Affecte la distribution du Cu (augmentation du Cu dans le foie). |
|
Plomb (Pb) |
Modifie le métabolisme du Ca; un régime alimentaire carencé en Ca fait augmenter l�absorption du Pb inorganique et la toxicité du Pb. Modifie le métabolisme du Fe; un régime carencé en Fe accroît la toxicité du Pb, tandis que son influence sur l�absorption du Pb est équivoque. |
Note: les données sont obtenues principalement à partir d�études expérimentales chez le rat, les données cliniques et épidémiologiques pertinentes (en particulier sur les relations quantitatives dose-réponse) faisant généralement défaut (Elsenhans et coll., 1991; Fergusson,1990; Teli�man et coll., 1993).
On sait depuis longtemps que chaque individu réagit différemment aux produits chimiques présents dans l’environnement. L’explosion récente de la biologie moléculaire et de la génétique a favorisé une meilleure compréhension des causes moléculaires d’une telle variabilité. Les facteurs déterminants de cette réponse individuelle aux produits chimiques incluent les différences importantes existant au niveau d’une dizaine de superfamilles d’enzymes, baptisées enzymes métabolisant les xénobiotiques (étrangers à l’organisme) ou enzymes métabolisant les médicaments. Bien qu’on leur ait longtemps prêté un pouvoir détoxifiant, on sait maintenant que ces enzymes sont également capables de transformer de nombreux composés inertes en intermédiaires fortement toxiques. On a pu identifier récemment, au niveau des gènes codant pour ces enzymes, des différences légères ou importantes, responsables de disparités marquées de leur activité enzymatique. Il est maintenant établi que chaque individu possède un effectif d’activités enzymatiques métabolisant les xénobiotiques qui lui est propre, cette diversité pouvant être assimilée à une «empreinte métabolique». C’est l’interaction complexe de ces nombreuses superfamilles d’enzymes qui détermine non seulement le devenir d’un produit chimique chez un individu donné et son potentiel de toxicité, mais également l’évaluation de l’exposition. Dans cet article, nous avons choisi la superfamille des enzymes du cytochrome P450 pour illustrer les remarquables progrès accomplis dans la compréhension de la réponse individuelle aux produits chimiques. Le développement de tests ADN relativement simples identifiant les altérations des gènes spécifiques de ces enzymes permet maintenant de prévoir de façon plus exacte la réponse individuelle à l’exposition à un produit chimique. On peut espérer que ces tests conduiront au développement de la toxicologie préventive. Chaque individu pourrait ainsi connaître les produits chimiques auxquels il serait particulièrement sensible, ce qui lui éviterait de s’exposer à des produits dont on ne soupçonnait pas auparavant qu’ils aient chez lui des effets toxiques, voire cancérogènes.
L’être humain est exposé tous les jours, bien souvent à son insu, à une multitude de produits chimiques. Beaucoup de ces produits extrêmement toxiques proviennent de sources très diverses, environnementales ou alimentaires. La relation entre ces expositions et la santé humaine a été, et continue d’être, un des principaux objectifs de la recherche biomédicale dans le monde.
Les exemples de ce véritable bombardement de produits chimiques ne manquent pas. Plus de 400 agents ont été mis en évidence dans le vin rouge et identifiés. La cigarette ne produit pas moins de 1 000 espèces chimiques différentes et les produits cosmétiques et savons parfumés renferment un nombre incalculable de produits chimiques. Dans l’agriculture, la situation n’est pas différente: pour traiter les terres cultivées aux Etats-Unis, on utilise tous les ans plus de 75 000 agents chimiques sous forme de pesticides, d’herbicides et d’engrais; ces produits sont captés par les plantes et les herbivores, ou les poissons dans les eaux environnantes, avant d’être ingérés par l’être humain qui se trouve à la fin de la chaîne alimentaire. Cette liste ne serait pas complète sans qu’on y ajoute deux autres sources importantes de produits chimiques: a) les médicaments consommés de façon prolongée; b) les substances dangereuses auxquelles les travailleurs sont exposés au cours de la vie active.
Il est maintenant établi que l’exposition aux produits chimiques peut avoir de nombreux effets nocifs sur la santé humaine et entraîner l’apparition de maladies chroniques et de bien des cancers. Des travaux effectués ces dix dernières années ont permis de mieux comprendre la base moléculaire de ces relations. Ils ont aussi permis de prendre conscience des différences individuelles de sensibilité aux effets nocifs des produits chimiques.
Les efforts actuels pour prévoir la réponse de l’humain à une exposition à un produit chimique associent deux démarches fondamentales (voir figure 33.2): surveillance de l’exposition humaine à l’aide de marqueurs biologiques (biomarqueurs) et prévision de la réaction probable d’un individu à un niveau d’exposition donné. Ces deux approches, pour importantes qu’elles soient, n’en sont pas moins très différentes l’une de l’autre. Le présent article porte sur les facteurs génétiques à l’origine de la sensibilité individuelle à l’exposition aux produits chimiques. Ce champ de recherche porte le nom général d’écogénétique ou de pharmacogénétique (Kalow, 1962, 1992). Si des progrès ont été réalisés récemment dans la mise en évidence des sensibilités individuelles à une toxicité chimique, c’est qu’on comprend maintenant mieux les mécanismes qui permettent à l’humain et aux autres mammifères de détoxifier tous ces produits et qu’on a une meilleure connaissance de la remarquable complexité des systèmes enzymatiques entrant en jeu.
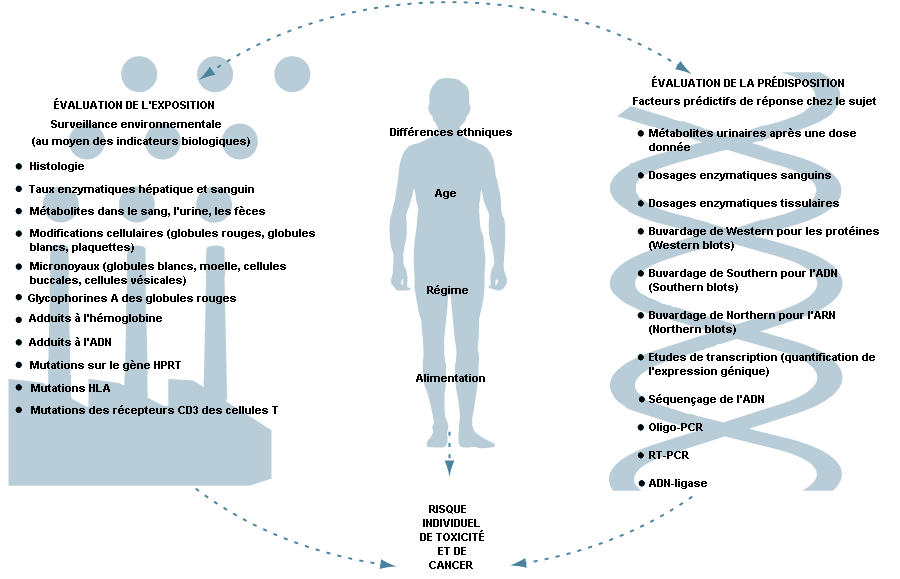
Nous expliquerons dans un premier temps comment les réponses toxiques varient d’un être humain à l’autre, avant de présenter quelques-unes des enzymes responsables d’une telle variabilité, liée à des différences de métabolisme des xénobiotiques. Ensuite, nous dresserons l’historique et la nomenclature de la superfamille des cytochromes P450 et nous décrirons brièvement cinq polymorphismes du P450 humain et plusieurs polymorphismes non liés au P450, responsables de différences de réponse toxique chez l’humain. Puis, à la lumière d’un exemple, nous montrerons comment les différences génétiques individuelles peuvent se répercuter sur l’exposition établie par contrôle d’ambiance. En dernier lieu, nous traiterons du rôle des enzymes métabolisant les xénobiotiques au niveau de fonctions vitales critiques.
Les toxicologues et les pharmacologues ont coutume de parler de DL50, de DMA50 et de DE50 pour désigner respectivement, dans le cas d’un médicament donné, la dose létale, la dose maximale admissible et la dose efficace dans 50% d’une population. On peut se demander ce que ces doses signifient pour chacun de nous en tant qu’individu. Ce qu’on entend par là, c’est qu’un individu très sensible peut être 500 fois plus affecté ou avoir 500 fois plus de chances d’être affecté que le sujet le plus résistant d’une population donnée; dans l’absolu, les valeurs de DL50 (et de DMT50 ou de DE50) ne veulent pas dire grand-chose, elles n’ont un sens qu’en référence à une population.
La figure 33.3 illustre la relation dose-réponse hypothétique observée chez des individus appartenant à une population donnée exposée à un toxique. Ce diagramme général pourrait représenter un carcinome bronchique en fonction du nombre de cigarettes fumées, une acné chlorique par rapport à la concentration de dioxine présente sur le lieu de travail, un asthme en fonction des concentrations de l’air en ozone ou en aldéhyde, un érythème solaire par rapport aux rayons ultraviolets, la diminution du temps de coagulation en fonction de la consommation d’aspirine, ou encore la souffrance gastro-intestinale en réaction à la quantité de poivre jalapeño consommée. Généralement, pour chacun de ces cas, plus l’exposition est importante, plus la réponse toxique est intense. La majorité de la population présentera une réponse toxique moyenne avec son écart-type en fonction de la dose. Le sujet «hyperrésistant» (en bas à droite dans la figure 33.3) est celui qui répond le moins aux expositions ou aux doses plus élevées, le sujet «hypersensible» (en haut à gauche) étant celui qui répond de façon excessive à une exposition ou à une dose relativement faibles. Ces sujets hors norme, réagissant de manière très différente par rapport à la majorité des individus de la population, représentent des variants génétiques importants qui peuvent aider les scientifiques à comprendre les mécanismes moléculaires sous-jacents responsables de la réponse toxique.
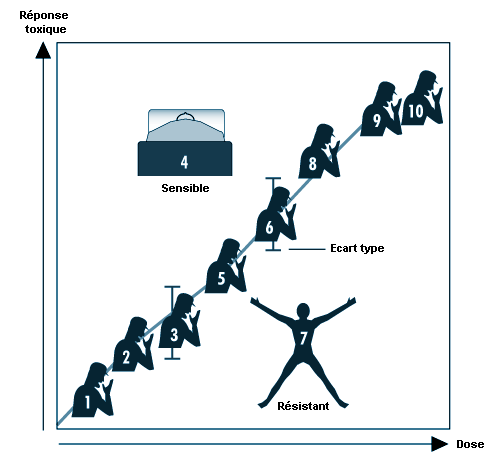
Grâce à ces sujets d’exception et dans le cadre d’études familiales, les scientifiques d’un certain nombre de laboratoires ont pris conscience de l’importance de l’hérédité mendélienne dans la réponse à un toxique donné. Ils ont ensuite pu faire appel à la biologie moléculaire et à la génétique pour mettre en évidence le mécanisme sous-jacent au niveau génétique (génotype) responsable de l’affection provoquée par l’environnement (phénotype).
Comment l’organisme répond-il à la multitude de produits chimiques exogènes auxquels il est exposé? L’être humain et les autres mammifères ont développé des systèmes enzymatiques très complexes comportant plus d’une dizaine de superfamilles d’enzymes. Tous les produits chimiques auxquels l’humain est exposé ou presque subissent une modification enzymatique qui favorise ensuite leur élimination en dehors de l’organisme. Ces enzymes sont souvent appelées de façon globale enzymes métabolisant les médicaments ou enzymes métabolisant les xénobiotiques. En fait, l’un comme l’autre de ces termes sont impropres et ce pour deux raisons. Premièrement, un bon nombre de ces enzymes métabolisent non seulement les médicaments, mais aussi des centaines de milliers de produits chimiques présents dans l’environnement et dans les aliments. Deuxièmement, toutes ces enzymes ont également comme substrat des composés endogènes normaux et aucune d’entre elles ne métabolise uniquement des produits chimiques étrangers.
Depuis plus de quatre décennies, les processus métaboliques (à médiation enzymatique) dépendant de ces enzymes sont classés en réactions de phase I ou de phase II (voir figure 33.4). Les réactions de phase I («fonctionnalisation») correspondent généralement à des modifications structurales assez peu importantes du produit chimique parent par des réactions d’oxydation, de réduction ou d’hydrolyse permettant l’obtention d’un métabolite plus hydrosoluble. Les réactions de phase I fournissent une «clé» sans laquelle les modifications ultérieures du composé par les réactions de phase II ne peuvent pas se faire. Les réactions de phase I sont principalement gérées par une superfamille d’enzymes hautement polyvalentes, appelées cytochromes P450, bien que d’autres superfamilles d’enzymes puissent également intervenir (voir figure 33.5).
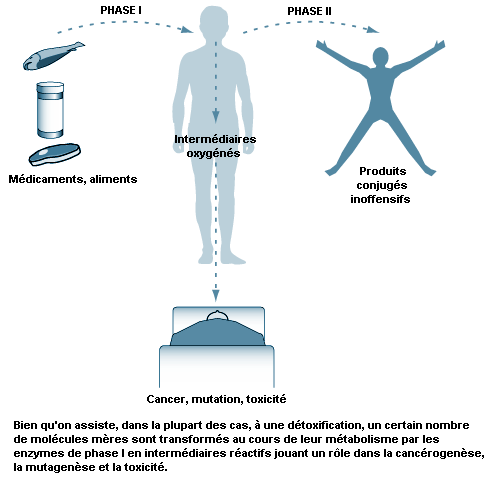
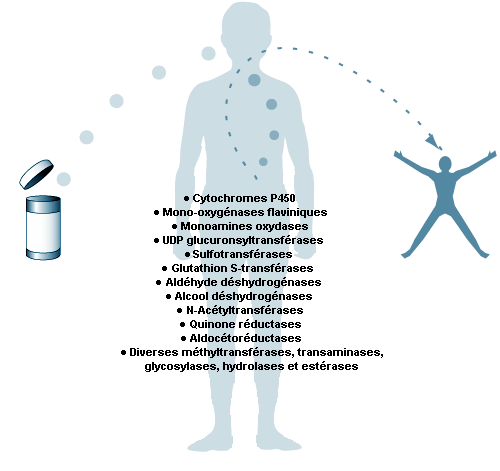
Les réactions de phase II supposent le couplage d’une molécule endogène hydrosoluble à un produit chimique (produit chimique initial ou métabolite de phase I) afin d’en faciliter l’excrétion. On appelle souvent les réactions de phase II réactions de «conjugaison» ou de «dérivation». Les superfamilles enzymatiques catalysant les réactions de phase II sont habituellement désignées selon le type de molécule endogène impliquée dans la réaction de conjugaison: acétylation par les N-acétyltransférases, sulfoconjugaison par les sulfotransférases, conjugaison du glutathion par les glutathion-transférases et glucuronidation par les UDP glucuronosyltransférases (voir figure 33.5). Le foie est le principal organe du métabolisme des xénobiotiques, bien qu’on trouve aussi certaines enzymes participant à leur métabolisme à un taux assez élevé dans le tractus gastro-intestinal, les gonades, le poumon, le cerveau et les reins, ainsi qu’en plus ou moins grande quantité dans toute cellule vivante.
Avec le progrès des connaissances sur les processus chimiques et biologiques conduisant à des manifestations pathologiques, on s’est peu à peu rendu compte que les enzymes métabolisant les xénobiotiques ont un mode de fonctionnement ambivalent (voir figure 33.4). Le plus souvent, les produits chimiques liposolubles sont transformés en métabolites hydrosolubles plus faciles à excréter. Cependant, il arrive aussi que ces mêmes enzymes transforment des produits chimiques inertes en molécules intermédiaires hautement réactives. Ces intermédiaires peuvent réagir avec des macromolécules cellulaires telles que les protéines et l’ADN. Ainsi, pour chaque produit chimique auquel l’être humain est exposé, deux voies compétitives potentielles coexistent, celle de l’activation métabolique et celle de la détoxification.
En génétique humaine, chaque gène (locus) est localisé sur l’une des 23 paires de chromosomes. Les deux allèles (un présent sur chaque chromosome de la paire) peuvent être identiques, mais ils peuvent aussi être différents l’un de l’autre. Par exemple, les allèles B et b, où B (yeux marron) est dominant par rapport à b (yeux bleus): les individus de phénotype yeux marron peuvent avoir comme génotype soit BB soit Bb, alors que les individus de phénotype yeux bleus peuvent seulement avoir le génotype bb.
Un polymorphisme correspond à la présence de deux ou de plusieurs phénotypes héréditaires stables (traits) ayant pour origine le(s) même(s) gène(s) qui se maintiennent dans la population, souvent pour des raisons peu évidentes. Pour qu’un gène soit polymorphe, il faut que son produit ne soit pas essentiel au développement, à la reproduction ou à d’autres processus vitaux critiques. En fait, on a pris l’habitude d’expliquer par ce phénomène de «polymorphisme équilibré», où l’hétérozygote a un avantage net de survie sur l’homozygote (par exemple, résistance à la malaria et allèle de l’hémoglobine drépanocytaire), la présence dans la population d’un allèle dont la fréquence élevée serait autrement inexplicable (Gonzalez et Nebert, 1990).
Les différences génétiques du métabolisme des médicaments et produits chimiques environnementaux sont connues depuis plus d’une quarantaine d’années (Kalow, 1962, 1992). Ces différences sont souvent appelées polymorphismes pharmacogénétiques ou, de façon plus générale, écogénétiques. Ces polymorphismes représentent des allèles variants survenant à des fréquences relativement élevées dans la population et généralement associés à des aberrations d’expression ou de fonction enzymatique. Par le passé, les polymorphismes ont bien souvent été mis en évidence à la suite de réponses inattendues à des agents thérapeutiques. Plus récemment, la technologie de l’ADN recombinant a permis aux scientifiques d’identifier les altérations génétiques précises responsables de certains polymorphismes. Ces polymorphismes sont maintenant caractérisés pour de nombreuses enzymes du métabolisme des xénobiotiques, y compris celles de phase I et de phase II. Au fur et à mesure qu’on découvre de nouveaux polymorphismes, on s’aperçoit que chaque individu possède un effectif distinct d’enzymes métabolisant les xénobiotiques. On peut dire de cette diversité qu’elle constitue son «empreinte métabolique» et que la réponse particulière de chaque individu à un produit chimique dépend de l’interaction complexe des superfamilles d’enzymes responsables du métabolisme des xénobiotiques (Kalow, 1962, 1992; Nebert, 1988; Gonzalez et Nebert, 1990; Nebert et Weber, 1990).
Comment améliorer la qualité des facteurs de prédiction des réponses humaines aux produits chimiques toxiques? Les progrès de nos connaissances sur les systèmes enzymatiques intervenant dans le métabolisme des médicaments doivent nous permettre de comprendre de manière précise quelles sont les enzymes qui déterminent le devenir métabolique de tout produit chimique. Les données recueillies à partir d’études expérimentales chez les rongeurs ont certainement fourni à cet égard des informations utiles. Cependant, des différences interespèces importantes au niveau des enzymes métabolisant les xénobiotiques appellent à la prudence dans l’extrapolation des données animales aux populations humaines. Pour pallier cette difficulté, de nombreux laboratoires ont mis au point des systèmes qui utilisent diverses lignées cellulaires en culture produisant des enzymes humaines fonctionnelles, stables et en forte concentration (Gonzalez, Crespi et Gelboin, 1991). De telles enzymes ont ainsi été produites à partir de bactéries, de levures, d’insectes ou de mammifères.
Afin de mieux préciser le métabolisme des produits chimiques, des systèmes multienzymatiques ont été également développés avec succès dans une lignée cellulaire unique (Gonzalez, Crespi et Gelboin, 1991). Ces lignées cellulaires fournissent des renseignements précieux sur les enzymes participant à la transformation métabolique d’un composé donné et des métabolites éventuellement toxiques. Si cette information peut être ensuite recoupée avec la présence et la concentration d’une enzyme au niveau des tissus humains, elle devrait permettre de prévoir une réponse de façon fiable.
La superfamille des cytochromes P450 est l’une des superfamilles d’enzymes du métabolisme des médicaments qui a été le plus étudiée, en raison des disparités individuelles importantes qui caractérisent la réponse aux produits chimiques. Le terme cytochrome P450 est une désignation générique commode, employée pour définir cette grande superfamille d’enzymes essentielles au métabolisme d’innombrables substrats endogènes et exogènes. Il a été utilisé la première fois en 1962 pour décrire un pigment cellulaire inconnu qui, une fois réduit et lié au monoxyde de carbone, a produit un pic d’absorption caractéristique à 450 nm. Depuis le début des années quatre-vingt, la technologie du clonage d’ADNc a permis d’avoir un remarquable aperçu de la multiplicité des enzymes du cytochrome P450. Actuellement, plus de 400 gènes distincts du cytochrome P450 ont été identifiés chez les animaux, les plantes, les bactéries et les levures. On estime que toute espèce de mammifères, l’humain par exemple, possède pas moins de 60 gènes différents de P450 (Nebert et Nelson, 1991). Cette multiplicité a exigé la mise au point d’une nomenclature normalisée (Nebert et coll., 1987; Nelson et coll., 1993). Proposé pour la première fois en 1987 et mis à jour deux fois par an, ce système de nomenclature est basé sur la comparaison des séquences d’amino-acides entre les protéines de cytochrome P450. Les gènes du P450 sont divisés en familles et en sous-familles: les enzymes à l’intérieur d’une famille et celles à l’intérieur d’une même sous-famille présentent respectivement une similitude en acides aminés de plus de 40% et de 55%. Les gènes du P450 sont caractérisés par un symbole commun, CYP, suivi d’un chiffre arabe désignant la famille P450, puis d’une lettre spécifiant la sous-famille et d’un autre chiffre arabe propre au gène individuel (Nelson et coll. 1993; Nebert et coll. 1991). Ainsi, CYP1A1 représente le gène P450 1 dans la famille 1, sous-famille A.
En février 1995, la base de données du cytochrome P450 comportait 403 gènes CYP, répartis en 59 familles et 105 sous-familles. Les familles sont au nombre de 8 pour les eucaryotes inférieurs, 15 pour les plantes et 19 pour les bactéries. Les 15 familles de gènes P450 humains comprennent 26 sous-familles; 22 d’entre elles ont été localisées au niveau chromosomique. Certaines séquences sont nettement orthologues pour de nombreuses espèces: un gène CYP17 (stéroïde 17α-hydroxylase) a été identifié chez tous les vertébrés examinés jusqu’à ce jour; d’autres séquences à l’intérieur d’une sous-famille sont fortement dupliquées, ce qui rend l’identification des paires orthologues impossible (cas de la sous-famille CYP2C). Curieusement, l’humain et la levure partagent un gène orthologue dans la famille CYP51. Pour les lecteurs cherchant des informations complémentaires sur la superfamille des cytochromes P450, il existe de nombreux ouvrages très bien documentés (Nelson et coll., 1993; Nebert et coll., 1991; Nebert et McKinnon, 1994; Guengerich 1993; Gonzalez 1992).
Le succès du système de nomenclature des P450 a entraîné le développement de systèmes terminologiques semblables pour les UDP glucuronosyltransférases (Burchell et coll., 1991) et les mono-oxygénases à flavine (Lawton et coll., 1994). Des systèmes de nomenclature similaires sont également en cours de développement pour d’autres superfamilles d’enzymes métabolisant les médicaments (sulfotransférases, époxyde hydrolases et aldéhyde déshydrogénases, par exemple).
Récemment, la superfamille des gènes P450 des mammifères a été divisée en trois groupes (Nebert et McKinnon, 1994): ceux qui interviennent surtout dans le métabolisme des xénobiotiques et ceux qui participent à la synthèse de diverses hormones stéroïdes d’une part, et à d’autres fonctions endogènes importantes, de l’autre. Ce sont les enzymes du P450 métabolisant les xénobiotiques qui sont les plus intéressantes sur le plan de la toxicité.
Les enzymes P450 intervenant dans le métabolisme des produits exogènes et des médicaments sont presque toujours présentes dans les familles CYP1, CYP2, CYP3 et CYP4. Ces enzymes P450 catalysent de nombreuses réactions métaboliques, un seul cytochrome P450 étant souvent capable de métaboliser plusieurs composés différents. De plus, des enzymes P450 multiples peuvent métaboliser un composé sur différents sites. Un composé peut également être métabolisé au niveau d’un site unique par plusieurs P450, mais à des vitesses variables.
Les enzymes P450 métabolisant les médicaments sont dotés d’une propriété importante: beaucoup de ces gènes sont inductibles par les substances mêmes qui leur servent de substrat. Mais d’autres enzymes P450 sont induites par des molécules non substrat. Ce phénomène d’induction enzymatique est à la base de nombreuses interactions médicamenteuses, importantes du point de vue thérapeutique.
Bien que présentes dans de nombreux tissus, c’est dans le foie, principal site du métabolisme des médicaments, qu’on retrouve ces enzymes P450 à des taux relativement élevés. Certaines enzymes du P450 métabolisant les xénobiotiques possèdent une activité vis-à-vis de plusieurs substrats endogènes (acide arachidonique, par exemple). Cependant, on estime en général que la plupart des enzymes P450 métabolisant les xénobiotiques ne jouent pas un rôle physiologique important, bien qu’on ne l’ait pas encore établi expérimentalement. La rupture sélective de l’homozygotie, ou «knock-out», de certains gènes P450 métabolisant les xénobiotiques chez la souris permettra d’obtenir dans un avenir proche des informations sans équivoque sur le rôle physiologique des P450 métabolisant les xénobiotiques (pour une synthèse sur le ciblage des gènes, on peut se reporter à l’ouvrage de Capecchi, 1994).
Par opposition aux familles de P450 codant pour des enzymes impliquées principalement dans des processus physiologiques, les familles codant pour des enzymes P450 métabolisant les xénobiotiques manifestent une spécificité d’espèce marquée et comportent fréquemment de nombreux gènes actifs dans chaque sous-famille (Nelson et coll., 1993; Nebert et coll., 1991). Etant donné leur manque apparent de substrats physiologiques, il est possible que les enzymes P450 des familles CYP1, CYP2, CYP3 et CYP4, apparues il y a quelques centaines de millions d’années, aient évolué afin de détoxifier les produits chimiques étrangers rencontrés dans l’environnement et dans l’alimentation. En clair, l’évolution des P450 métabolisant les xénobiotiques se serait déroulée à une période bien antérieure à celle de la synthèse de la plupart des produits chimiques synthétiques auxquels l’humain est maintenant exposé. Les gènes de ces quatre familles ont vraisemblablement évolué et divergé selon les espèces en raison de leur exposition aux métabolites des végétaux au cours des 1,2 milliard d’années passées. Cette «guerre animal-plante» (Gonzalez et Nebert, 1990), comme on l’appelle de façon imagée, est un phénomène au cours duquel les plantes ont développé de nouveaux produits chimiques (phytoalexines) comme mécanisme de défense pour ne pas être ingérées par les animaux; les animaux ont réagi à ce changement en se dotant de nouveaux gènes P450 pour s’adapter à la diversification des substrats. Les exemples décrits récemment de guerre chimique plante-insecte et plante-champignon impliquant une détoxification de substrats toxiques par les P450 donnent plus de poids à cette explication (Nebert, 1994). Nous consacrons les quelques pages qui suivent à plusieurs polymorphismes humains des enzymes P450 métabolisant les xénobiotiques dans lesquels les déterminants génétiques de la réponse toxique semblent avoir une importance capitale. Jusqu’à tout récemment, on soupçonnait l’existence d’un polymorphisme P450 lorsqu’on observait une variance inattendue dans la réponse de patients à un agent thérapeutique. C’est ainsi que plusieurs polymorphismes portent le nom du médicament qui a permis à l’origine de les identifier. Plus récemment, les efforts de recherche ont surtout porté sur l’identification précise des enzymes P450 impliquées dans le métabolisme de produits chimiques pour lesquels une variance est observée et sur la caractérisation exacte des gènes P450 concernés. Comme nous l’avons mentionné précédemment, l’activité mesurable d’une enzyme P450 vis-à-vis d’un produit chimique type porte le nom de phénotype. Les différences alléliques du gène P450 pour chaque individu sont appelées génotype P450. Avec l’étude de plus en plus minutieuse des gènes P450, la base moléculaire précise de la variance phénotypique précédemment décrite devient plus claire.
La sous-famille CYP1A comprend deux enzymes chez l’humain et tous les autres mammifères. Ces enzymes, appelées CYP1A1 et CYP1A2 dans la nomenclature officielle, revêtent un intérêt considérable, car elles participent à l’activation métabolique de nombreux procancérogènes et sont également induites par plusieurs composés posant des problèmes sur le plan toxicologique, dont la dioxine. Par exemple, la CYP1A1 active, en les métabolisant, de nombreux composés qu’on retrouve dans la fumée de cigarette. De même, la CYP1A2 active de nombreuses arylamines utilisées dans l’industrie des colorants et associées au cancer de la vessie. Elle active également le 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), une nitrosamine dérivée du tabac. Les CYP1A1 et CYP1A2, qui sont présentes à des taux très élevés dans les poumons des fumeurs de cigarettes, sont produites par les hydrocarbures polycycliques présents dans la fumée. Les niveaux d’activité de ces deux enzymes sont donc considérés comme des facteurs importants de la réaction individuelle à de nombreux produits chimiques potentiellement toxiques.
La sous-famille CYP1A a vu son intérêt toxicologique fortement augmenter à la suite d’un rapport paru en 1973 établissant une corrélation entre l’induction de la CYP1A1 chez les fumeurs et la prédisposition individuelle au cancer pulmonaire (Kellermann, Shaw et Luyten-Kellermann, 1973). De nombreux laboratoires se sont alors intéressés aux mécanismes moléculaires de l’induction des CYP1A1 et CYP1A2. Le processus d’induction se fait par l’intermédiaire d’une protéine appelée récepteur Ah à laquelle se fixent les dioxines et les produits chimiques structurellement apparentés. Le nom Ah est dû au caractère aryl hydrocarboné de nombreux inducteurs du CYP1A. Précisons que, selon la souche de souris, les différences au niveau du gène codant pour le récepteur Ah entraînent des disparités notables dans la toxicité d’un produit chimique et dans la réponse à cette toxicité. Il semble exister chez l’humain un polymorphisme du gène du récepteur Ah: ainsi, environ un dixième de la population présente une induction importante du CYP1A1 susceptible de l’exposer à un plus grand risque de certains cancers induits chimiquement que les neuf autres dixièmes de la population. Le rôle que joue le récepteur Ah dans le contrôle des enzymes de la sous-famille du CYP1A et dans la réponse humaine à une exposition chimique a fait l’objet de plusieurs études bibliographiques récentes (Nebert, Petersen et Puga, 1991; Nebert, Puga et Vasiliou, 1993).
Existe-t-il d’autres polymorphismes pouvant déterminer le taux protéique de CYP1A dans la cellule? Un polymorphisme du gène CYP1A1, identifié chez les Japonais fumeurs de cigarettes, semble avoir une incidence sur le risque de cancer pulmonaire, bien que ce même polymorphisme ne paraisse pas exercer d’influence sur ce risque dans d’autres groupes ethniques (Nebert et McKinnon, 1994).
On sait depuis longtemps que les individus ne métabolisent pas tous à la même vitesse le médicament anticonvulsivant (S)-méphénytoïne (Guengerich, 1989). A cause d’une déficience, entre 2 et 5% des Caucasiens et jusqu’à 25% des Asiatiques présentent un plus grand risque de réaction toxique à ce médicament. On sait depuis longtemps aussi que cette anomalie enzymatique est due à un membre de la sous-famille CYP2C; cependant, la base moléculaire précise de cette déficience a donné lieu à de nombreuses controverses, la sous-famille CYP2C ne contenant pas moins de six gènes. Récemment, on a pu démontrer que la principale cause de cette déficience était la mutation d’une seule base au niveau du gène CYP2C19 (Goldstein et de Morais, 1994). Un simple test ADN, par réaction en chaîne de la polymérase, a également été mis au point pour identifier rapidement cette mutation dans des populations humaines (Goldstein et de Morais, 1994).
La variation sans doute la mieux caractérisée d’un gène P450 est celle du CYP2D6. Plus d’une dizaine de cas de mutations, de réarrangements et de délétions touchant ce gène ont été décrits (Meyer, 1994). L’existence de ce polymorphisme a été soupçonnée pour la première fois il y a une vingtaine d’années lorsqu’on s’est aperçu que les patients traités par la débrisoquine, médicament antihypertenseur, ne présentaient pas tous la même réponse clinique. C’est de cette époque que date l’expression polymorphisme de la débrisoquine pour désigner les altérations du gène CYP2D6 à l’origine des variations de l’activité enzymatique.
Avant les études sur l’ADN, on classait les individus en métaboliseurs lents ou rapides de la débrisoquine en se fondant sur les concentrations de métabolites dans les échantillons urinaires. On sait maintenant que les altérations du gène CYP2D6 permettent non seulement de distinguer les individus qui métabolisent lentement ou rapidement la débrisoquine, mais aussi ceux qui ont un métabolisme ultrarapide. La plupart des altérations du gène CYP2D6 sont associées à une déficience partielle ou totale de l’enzyme; cependant, on a constaté récemment qu’il existait des sujets de deux familles possédant de multiples copies fonctionnelles du gène CYP2D6, altération à l’origine d’un métabolisme ultrarapide des substrats du CYP2D6 (Meyer, 1994). Cette observation remarquable donne un nouvel éclairage à la question du large spectre de l’activité du CYP2D6 précédemment constatée dans des études de population. Les altérations de fonction du CYP2D6 revêtent un intérêt tout particulier, étant donné que plus de 30 médicaments prescrits de façon courante sont métabolisés par cette enzyme. Le statut individuel du CYP2D6 est donc un facteur déterminant des réponses thérapeutiques et toxiques après administration d’une médication. Un débat récent a d’ailleurs eu lieu sur la nécessité de prendre en considération ce statut pour l’administration en toute sécurité de médicaments psychiatriques ou cardio-vasculaires à des patients.
Le rôle du polymorphisme du CYP2D6 dans la prédisposition individuelle à des maladies humaines telles que le cancer pulmonaire ou la maladie de Parkinson a fait l’objet d’études approfondies (Nebert et McKinnon, 1994; Meyer, 1994). Bien qu’il soit difficile d’en tirer des conclusions étant donné la diversité des protocoles employés, la majorité des études semblent indiquer une association entre les métaboliseurs rapides de la débrisoquine et le cancer pulmonaire. Pour l’heure, les raisons d’une telle association ne sont pas claires. Néanmoins, on a démontré que l’enzyme CYP2D6 métabolise la NNK, nitrosamine produite à partir du tabac.
L’amélioration des techniques d’étude de l’ADN permettra, tout en garantissant une évaluation plus précise du statut du CYP2D6, de préciser la relation entre le CYP2D6 et le risque pathogène. Alors que les métaboliseurs rapides paraissent plus sensibles à la survenue d’un cancer pulmonaire, pour une raison encore inconnue, les métaboliseurs lents semblent présenter une plus grande sensibilité à la maladie de Parkinson. Bien qu’il soit difficile de comparer ces études, il apparaît que les sujets métaboliseurs lents présentent de 2 à 2,5 fois plus de risques de développer une maladie de Parkinson.
Le gène CYP2E1 code pour une enzyme qui métabolise de nombreux produits chimiques, en particulier des médicaments et de nombreux cancérogènes de faible poids moléculaire. Cette enzyme, fortement inductible par l’alcool, peut jouer un rôle dans les lésions hépatiques produites par des produits chimiques tels que le chloroforme, le chlorure de vinyle ou le tétrachlorure de carbone. On la trouve surtout dans le foie, à un taux qui fluctue sensiblement d’un individu à l’autre. L’étude approfondie du gène CYP2E1 a permis d’identifier plusieurs polymorphismes (Nebert et McKinnon, 1994). La relation entre la présence de certaines variations structurelles du gène CYP2E1 et un risque apparemment plus faible de cancer pulmonaire a été établie dans certaines études; cependant, l’existence de différences interethniques marquées exige d’être approfondie pour pouvoir confirmer cette relation éventuelle.
Chez l’humain, quatre enzymes ont été identifiées dans la sous-famille CYP3A en raison de la similitude de leur séquence en acides aminés. Les enzymes du CYP3A métabolisent de nombreux médicaments couramment prescrits comme l’érythromycine et la cyclosporine. L’aflatoxine B1, contaminant cancérogène des produits alimentaires, est également un substrat du CYP3A. L’un des membres de la sous-famille humaine du CYP3A, appelé CYP3A4, est le principal composant de la famille des cytochromes P450 du foie humain; il est également présent au niveau du tractus gastro-intestinal. Comme c’est le cas pour de nombreuses enzymes du cytochrome P450, le taux de CYP3A4 est très variable selon les individus. Une seconde enzyme, CYP3A5, est présente dans le foie de 25% seulement des individus; la cause génétique de ces variations est inconnue. L’importance de la variabilité du CYP3A4 ou du CYP3A5 comme facteur génétique déterminant d’une réponse toxique n’a pas encore été établie (Nebert et McKinnon, 1994).
De nombreux polymorphismes existent également dans d’autres superfamilles d’enzymes métabolisant les xénobiotiques (glutathion transférases, UDP glucuronosyltransférases, p-oxonases, déshydrogénases, N-acétyltransférases ou mono-oxygénases à flavine, etc.). Etant donné que la toxicité finale d’un intermédiaire généré par le cytochrome P450 dépend de l’efficacité des réactions ultérieures de détoxification lors de la phase II, le rôle combiné des multiples polymorphismes enzymatiques revêt une importance capitale pour la prédisposition aux maladies provoquées par des produits chimiques. L’équilibre métabolique entre les réactions de phase I et de phase II (voir figure 33.4) joue donc selon toute vraisemblance un rôle clé dans la survenue de ce type de pathologies chez l’humain et constitue le facteur génétique déterminant d’une réponse toxique.
Un exemple bien étudié de polymorphisme d’une enzyme de phase II est celui d’un membre de la superfamille des enzymes glutathion S-transférases, la GST mu ou GSTM1. Cette enzyme particulière a un intérêt toxicologique considérable, puisqu’elle paraît participer à la détoxification des métabolites toxiques produits par l’enzyme CYP1A1 à partir de la fumée de cigarette. Le polymorphisme du gène de la glutathion transférase consiste en une absence totale d’enzyme fonctionnelle chez la moitié des Caucasiens. Cette absence d’enzyme de phase II semble associée à une prédisposition accrue au cancer pulmonaire. En groupant les individus en fonction des variants du gène CYP1A1 et de la délétion ou de la présence d’un gène GSTM1 fonctionnel, on a pu démontrer que le risque de cancer pulmonaire dû au tabagisme varie considérablement (Kawajiri, Watanabe et Hayashi, 1994). En particulier, les sujets présentant une altération rare du gène CYP1A1, associée à l’absence de gène GSTM1, ont un risque accru (neuf fois plus élevé) de développer un cancer pulmonaire lorsqu’ils sont exposés à une concentration pourtant relativement faible de fumée de cigarette. Précisons que des différences interethniques semblent intervenir dans ces gènes variants et qu’il serait opportun d’effectuer des études complémentaires pour préciser le rôle de telles altérations en pathologie (Kalow, 1962; Nebert et McKinnon, 1994; Kawajiri, Watanabe et Hayashi, 1994).
La réponse toxique à un agent environnemental peut être très fortement augmentée lorsqu’un même individu présente deux anomalies pharmacogénétiques comme la combinaison d’un polymorphisme de la N-acétyltransférase (NAT2) et d’un polymorphisme de la glucose-6-phosphate déshydrogénase (G6PD).
L’exposition professionnelle aux arylamines constitue un risque important de cancer de la vessie. Depuis les études remarquables de Cartwright en 1954, on sait que le statut des N-acétyleurs est un facteur étiologique du cancer de la vessie induit par les colorants azoïques. Il existe une corrélation très significative entre le phénotype acétyleur lent, l’existence de cancer de la vessie et le degré d’invasion de ce cancer dans la paroi vésicale. A l’inverse, on a pu mettre en évidence une association significative entre le phénotype acétyleur rapide et l’incidence de carcinome côlo-rectal. Les gènes des N-acétyltransférases (NAT1, NAT2) ont été clonés et séquencés et, grâce aux techniques ADN, on est maintenant en mesure de détecter plus d’une dizaine de variants alléliques rendant compte du phénotype acétyleur lent. Le gène NAT2 est un gène polymorphe, surtout responsable de la variabilité de la réponse toxique aux produits chimiques présents dans l’environnement (Weber, 1987; Grant, 1993).
La glucose-6-phosphate déshydrogénase (G6PD) est une enzyme critique pour la production et le maintien du NADPH, sa faible activité ou son absence d’activité pouvant conduire à une hémolyse sévère induite par des médicaments ou des xénobiotiques due à une mauvaise concentration du glutathion réduit (GSH) dans les hématies. Le déficit en G6PD affecte au moins 300 millions de personnes dans le monde. Plus de 10% des Afro-Américains de sexe masculin présentent le phénotype le moins sévère, le «type méditerranéen» le plus sévère se retrouvant à une fréquence élevée (un sujet sur trois) dans certaines communautés sardes. Le gène G6PD a été cloné et localisé sur le chromosome X et de nombreuses mutations ponctuelles expliquent qu’on constate un tel degré d’hétérogénéité phénotypique chez les individus souffrant d’un tel déficit (Beutler, 1992).
Le thiozalsulfone, médicament de type sulfarylamine, est à l’origine d’une anémie hémolytique qui se manifeste par distribution bimodale dans la population à qui on l’administre. Lorsqu’ils sont traités par certains médicaments, les individus présentant à la fois un déficit en G6PD et le phénotype acétyleur lent sont plus affectés que ceux ayant seulement une carence en G6PD ou un phénotype acétyleur lent. Les acétyleurs lents déficitaires en G6PD sont au moins 40 fois plus sensibles à l’hémolyse induite par le thiozalsulfone que les acétyleurs rapides à statut normal en G6PD.
Pour assurer l’évaluation de l’exposition et la surveillance biologique (voir figure 33.2), il faut aussi disposer d’informations sur la constitution génétique de chaque individu. Pour une même exposition à un produit chimique dangereux, le taux des adduits à l’hémoglobine (ou d’autres marqueurs biologiques) peut varier de deux à trois ordres de grandeur d’un individu à l’autre, selon l’empreinte métabolique individuelle.
Des études semblables de pharmacogénétique ont été effectuées chez des travailleurs de l’industrie chimique en Allemagne (voir tableau 33.3). Par rapport aux autres phénotypes pharmaco-génétiques combinés possibles, les adduits à l’hémoglobine parmi les sujets exposés à l’aniline et à l’acétanilide sont de loin les plus élevés chez les acétyleurs lents présentant un déficit en G6PD. Cette étude a d’importantes implications sur le plan de l’évaluation de l’exposition. Il peut en effet arriver, compte tenu de la prédisposition génétique de chacun, que l’on sous-estime de deux ou de plusieurs ordres de grandeur l’exposition que subissent réellement deux individus exposés à une même concentration de produit chimique dangereux sur leur lieu de travail (si l’on en juge par des marqueurs biologiques tels que les adduits à l’hémoglobine). De même, le risque résultant d’un effet nocif pour la santé peut varier de deux ou plusieurs ordres de grandeur.
|
Type d�acétyleur |
Déficit en G6PD |
|||
|
Rapide |
Lent |
Non |
Oui |
Adduits à Hb |
|
+ |
+ |
2 |
||
|
+ |
+ |
30 |
||
|
+ |
+ |
20 |
||
|
+ |
+ |
100 |
||
Source: d�après Lewalter et Korallus, 1985.
Les observations que nous venons de faire pour le métabolisme valent aussi pour la liaison. Les différences héréditaires qui peuvent exister au niveau de la liaison d’agents environnementaux affectent de façon sensible la réponse toxique. Ainsi, les différences au niveau du gène cdm de la souris modifient fortement la sensibilité individuelle à la nécrose testiculaire induite par le cadmium (Taylor, Heiniger et Meier, 1973). Les différences d’affinité de liaison au récepteur Ah ont probablement une incidence sur la toxicité en cas d’expositions à la dioxine et sur la survenue du cancer (Nebert, Petersen et Puga, 1991; Nebert, Puga et Vasiliou, 1993).
La figure 33.6 résume le rôle du métabolisme et de la liaison dans la toxicité et le cancer. Les agents toxiques, tels qu’ils sont présents dans l’environnement, ou en raison de leur métabolisme ou de leur liaison, produisent des effets soit par voie génotoxique (apparition de lésions au niveau de l’ADN), soit par voie non génotoxique (absence de mutagenèse ou de lésions au niveau de l’ADN). Fait intéressant, on a établi récemment que les agents génotoxiques classiques peuvent agir par l’intermédiaire d’un signal de transduction non génotoxique dépendant du glutathion réduit (GSH), initié au niveau de la surface cellulaire ou dans son voisinage en absence d’ADN et hors du noyau cellulaire (Devary et coll., 1993). Cependant, les différences génétiques de métabolisme et de liaison restent les facteurs déterminants dans la façon dont les individus réagissent aux toxiques.
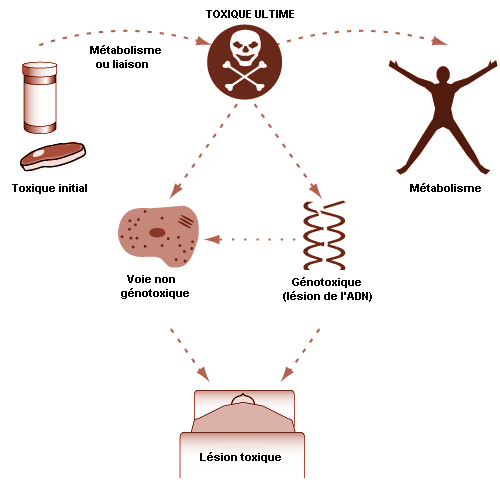
Les variations d’origine génétique du fonctionnement des enzymes métabolisant les médicaments revêtent une importance capitale dans la réponse individuelle aux produits chimiques. Ces enzymes sont essentielles pour comprendre le devenir et l’évolution d’un produit chimique étranger après une exposition.
Comme le montre la figure 33.6, l’importance des enzymes métabolisant les médicaments dans la sensibilité individuelle à l’exposition à un produit chimique est en fait beaucoup plus complexe que ne le laisse penser le présent exposé sur les xénobiotiques. Ainsi, au cours des deux dernières décennies, le rôle des mécanismes génotoxiques (mesure des adduits à l’ADN et aux protéines) est apparu clairement. On peut toutefois se demander si les mécanismes non génotoxiques ne sont pas aussi importants que les mécanismes génotoxiques dans la survenue des réponses toxiques.
Le rôle physiologique que de nombreuses enzymes métabolisant les médicaments jouent dans le métabolisme des xénobiotiques n’a pas, rappelons-le, été complètement élucidé. Selon Nebert (1994), les enzymes du métabolisme des médicaments qui sont présentes sur notre planète depuis plus de 3,5 milliards d’années étaient responsables à l’origine (et le sont encore, pour une part essentielle) de la régulation des niveaux cellulaires de nombreux ligands non peptidiques importants pour l’activation de la transcription de gènes en rapport avec la croissance, la différenciation, l’apoptose, l’homéostasie et les fonctions neuroendocriniennes. De plus, la toxicité de la plupart, si ce n’est de tous les agents environnementaux, s’exerce par l’intermédiaire d’une action agoniste ou antagoniste sur ces signaux de transduction (Nebert, 1994). Si l’on en croit cette hypothèse, la variabilité génétique des enzymes métabolisant les médicaments peut avoir des effets extrêmement graves sur de nombreux processus biochimiques essentiels pour la cellule et être de ce fait à l’origine d’importantes différences dans la réponse toxique. Le même scénario pourrait également expliquer de nombreuses réactions indésirables idiosyncrasiques rencontrées chez des patients prenant des médicaments prescrits de façon courante.
La dernière décennie a été marquée par des progrès remarquables dans la compréhension des bases génétiques de la réponse individuelle aux produits chimiques présents dans les médicaments, les aliments et les polluants environnementaux. Les enzymes métabolisant les médicaments ont une profonde influence sur la manière dont les individus réagissent aux produits chimiques. L’évolution de nos connaissances sur la multiplicité de ce type d’enzymes aidant, nous sommes chaque jour un peu mieux en mesure d’évaluer le risque toxique de nombreux médicaments et produits chimiques environnementaux. C’est le cas en particulier pour l’isoforme CYP2D6 du cytochrome P450. A l’aide de techniques ADN relativement simples, il est possible de prévoir la réponse probable à n’importe quel médicament métabolisé par cette enzyme; cette avancée est le gage d’une utilisation plus sûre de thérapeutiques indispensables, bien que potentiellement toxiques.
Au cours des années à venir, nous allons sans doute parvenir à identifier une multitude d’autres polymorphismes (phénotypes) impliquant des enzymes du métabolisme des médicaments. Ces informations seront le fruit de la mise en œuvre de techniques ADN améliorées, peu invasives, permettant d’identifier les génotypes dans la population humaine.
De telles études devraient être particulièrement précieuses pour évaluer le rôle des produits chimiques dans de nombreuses maladies environnementales d’origine encore inconnue. La prise en compte des multiples polymorphismes touchant les enzymes métabolisant les médicaments, agissant de manière combinée (voir tableau 33.3), représente à n’en pas douter un domaine de recherche particulièrement fertile. De telles études clarifieront la responsabilité des produits chimiques dans le développement du cancer. Au niveau collectif, cette information devrait permettre de prodiguer des conseils personnalisés aux individus afin de leur éviter de s’exposer aux produits chimiques susceptibles d’agir sur eux. Ce type d’information et de conseils, qui relève de la toxicologie préventive, nous aidera sans doute à faire face avec succès au nombre sans cesse croissant de produits chimiques auxquels nous sommes exposés.
La toxicologie mécanistique est l’étude du mécanisme par lequel les agents chimiques ou physiques réagissent avec les organismes vivants pour provoquer une réaction toxique. La connaissance du mécanisme toxique d’une substance améliore les possibilités de prévention et permet de concevoir des produits chimiques mieux tolérés. Elle constitue la base du traitement lors d’une surexposition, permet souvent d’éviter toute surexposition et garantit une meilleure compréhension des mécanismes biologiques fondamentaux. Dans la présente Encyclopédie, nous privilégions les expériences sur les animaux pour prédire les phénomènes toxiques chez l’humain. Les différents champs de la toxicologie — toxicologie mécanistique, descriptive, réglementaire, légale et environnementale (Klaassen, Amdur et Doull, 1991) — s’enrichissent grâce à la compréhension des mécanismes toxiques fondamentaux.
Comprendre le mécanisme du pouvoir toxique d’une substance permet d’améliorer les connaissances dans les différents domaines de la toxicologie. La connaissance des mécanismes d’action aide le législateur institutionnel à établir des limites de sécurité légales pour l’exposition humaine. Elle aide les toxicologues à recommander les moyens d’action nécessaires en vue d’assainir ou de réhabiliter les sites contaminés. Elle permet aussi, en se fondant sur les propriétés physico-chimiques d’une substance ou d’un mélange de substances, de choisir le type d’équipement de protection adapté aux besoins. Elle est également utile pour décider de la thérapie qui convient et pour développer de nouveaux médicaments permettant de traiter les pathologies humaines. Le mécanisme de toxicité permet souvent au toxicologue médico-légal de comprendre la façon dont un agent physique ou chimique peut causer le décès du sujet ou le rendre invalide.
Une fois le mécanisme de toxicité connu, la toxicologie descriptive permet de prévoir les effets toxiques des produits chimiques en question. Précisons, cependant, que l’absence d’information sur le mécanisme d’action ne devrait en aucun cas dissuader les professionnels de la santé de prendre les mesures de protection sanitaire qui s’imposent. Des décisions réfléchies sont arrêtées à partir de données chez l’animal et d’observations humaines pour établir des niveaux d’exposition sans danger. Traditionnellement, on s’assure une marge de sécurité en utilisant la «concentration sans effet nocif» ou la «concentration correspondant au plus faible effet nocif» observée dans des études sur des animaux de laboratoire (études par administrations répétées) et en divisant cette concentration par un facteur de 100 pour une exposition professionnelle ou de 1 000 pour les autres expositions humaines d’origine environnementale. Cette manière de procéder a de toute évidence porté ses fruits si l’on en croit le peu d’effets nocifs observés chez les travailleurs exposés à des concentrations de produits chimiques ne dépassant pas les valeurs limites établies. De plus, l’espérance de vie humaine continue d’augmenter, tout comme la qualité de vie. Dans l’ensemble, l’utilisation des données de la toxicologie a conduit à un contrôle efficace, qu’il soit réglementaire ou spontané. Le développement de la connaissance des mécanismes toxiques permettra d’accroître le caractère prévisionnel des modèles de risque actuels et d’améliorer sans cesse la prévention.
Complexe, la compréhension des mécanismes environnementaux suppose la connaissance de l’homéostasie (équilibre) des écosystèmes et des conditions entraînant la rupture de cet équilibre. Bien que ce sujet ne soit pas traité dans le présent article, il va de soi que les scientifiques seraient mieux en mesure de prendre des décisions judicieuses dans le domaine du traitement des déchets municipaux ou industriels s’ils comprenaient mieux les mécanismes toxiques et leurs conséquences dans un écosystème. La gestion des déchets, domaine de recherche en pleine expansion à l’heure actuelle, continuera d’être très important à l’avenir.
La majorité des études mécanistiques commence par une étude toxicologique descriptive chez l’animal ou par des observations cliniques chez l’humain. Idéalement, les études animales devraient comporter des observations cliniques et comportementales minutieuses, un examen biochimique attentif des paramètres sanguins ou urinaires pour rechercher des signes de dysfonctionnement des principaux systèmes biologiques de l’organisme ainsi qu’une étude histologique post mortem de tous les organes en vue de mettre en évidence des lésions (voir les tests recommandés par l’OCDE; les directives de la CE sur l’évaluation chimique; les directives de l’Agence américaine de protection de l’environnement (EPA); la réglementation japonaise sur les substances chimiques). On peut dire que cela correspondrait à un examen médical approfondi qui prendrait deux à trois jours en milieu hospitalier (à l’exception des examens post mortem).
La compréhension des mécanismes de toxicité repose sur l’art et la science de l’observation, la créativité dans la sélection de techniques pour valider les diverses hypothèses et l’intégration innovatrice des signes et des symptômes dans une relation de causalité. Lorsqu’on fait une étude mécanistique, on commence par étudier l’exposition à une substance, la distribution puis son devenir dans l’organisme (pharmacocinétique) en fonction du temps, avant de mettre en évidence l’effet toxique qui en résulte au niveau d’un système et en fonction de la dose. Les diverses substances peuvent entraîner des effets toxiques à différents niveaux du système biologique.
Dans les études mécanistiques, la voie d’exposition est généralement la même que lors de l’exposition humaine. Cette voie est importante car, en dehors des effets systémiques, on peut observer des effets locaux au niveau du site d’exposition, une fois que le produit chimique est passé dans le sang et s’est distribué dans l’organisme. Un exemple simple, mais convaincant, d’un effet local est l’irritation et, éventuellement, la corrosion cutanée après application de solutions de bases ou d’acides forts utilisées pour le nettoyage des surfaces dures. De même, l’irritation et la mort cellulaire peuvent s’observer au niveau des cellules nasales ou pulmonaires après exposition à des vapeurs ou à des gaz irritants tels que les oxydes d’azote ou l’ozone (les deux sont des polluants de l’air et se retrouvent parmi les composants du «smog» (brouillard chimique). Après son absorption par voie cutanée, pulmonaire ou gastro-intestinale et passage dans la circulation sanguine, le produit chimique peut être mis en évidence dans les organes ou les tissus à une concentration qui dépend de nombreux facteurs déterminant la pharmacocinétique du produit dans l’organisme. Le produit peut ensuite être aussi bien activé que détoxifié dans l’organisme comme indiqué ci-après.
La pharmacocinétique étudie l’absorption, la distribution, le métabolisme (modifications biochimiques dans l’organisme) et l’élimination ou l’excrétion du produit chimique en fonction du temps. Par rapport aux mécanismes de toxicité, ces variables pharmacocinétiques peuvent revêtir une très grande importance et, dans certains cas, être à l’origine de l’effet toxique ou de son absence. Si, par exemple, un produit est absorbé en quantité insuffisante, sa toxicité systémique (à l’intérieur de l’organisme) n’apparaîtra pas. Inversement, un produit chimique fortement réactif qui est détoxifié rapidement (en quelques secondes ou en quelques minutes) par les enzymes digestives ou hépatiques peut ne pas avoir le temps de manifester sa toxicité. Certains composés et mélanges halogénés polycycliques, de même que des métaux comme le plomb, n’entraînent pas de toxicité notable si leur excrétion est rapide; par contre, leur accumulation à une concentration suffisamment élevée provoque un effet toxique du fait de la lenteur de leur excrétion (parfois mesurée en années). Heureusement, la durée de rétention de la plupart des produits chimiques dans l’organisme n’est pas très longue. Même s’il s’accumule, un produit inoffensif n’aura pas d’effet toxique. La vitesse d’élimination en dehors de l’organisme ou de détoxification d’un produit chimique est souvent exprimée par sa demi-vie, qui correspond au temps nécessaire pour que 50% de ce produit soit excrété ou modifié en une forme non toxique.
Cependant, le fait qu’un produit chimique s’accumule dans une cellule ou un organe doit inciter à examiner attentivement son potentiel de toxicité dans l’organe en question. Plus récemment, des modèles mathématiques ont été élaborés pour extrapoler les variables pharmacocinétiques de l’animal à l’humain. Ces modèles pharmacocinétiques sont extrêmement utiles pour faire des hypothèses et vérifier si l’animal d’expérience peut servir de modèle prédictif pour l’humain. De nombreux articles et ouvrages ont été publiés à ce sujet (Gehring, Watanabe et Blau, 1976; Reitz, Nolan et Schumann, 1987; Nolan, Stott et Watanabe, 1995). La figure 33.7 montre un modèle pharmacocinétique simplifié.
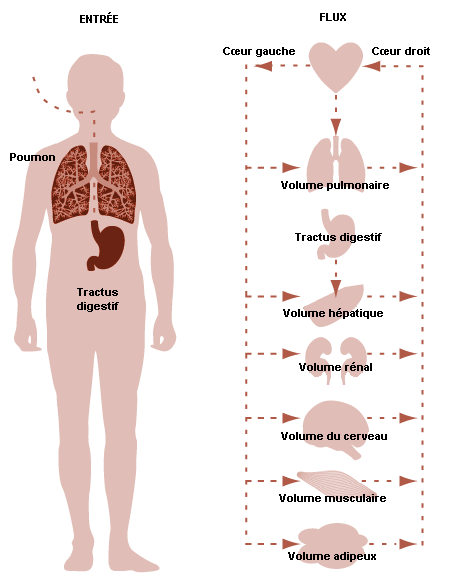
La toxicité peut s’exprimer à différents niveaux biologiques. La lésion peut être évaluée à l’échelle de la personne entière (ou de l’animal), de l’organe, de la cellule ou de la molécule. Par système, il faut entendre les systèmes ou appareils immunitaire, respiratoire, cardio-vasculaire, rénal, endocrinien, digestif, musculo-squelettique, sanguin, reproducteur et nerveux central. Les principaux organes sont le foie, les reins, les poumons, le cerveau, la peau, les yeux, le cœur, les testicules et les ovaires. Au niveau cellulaire ou biochimique, les effets nocifs peuvent perturber le métabolisme protéique normal ou les récepteurs endocriniens, inhiber le métabolisme énergétique ou encore entraver ou favoriser l’induction enzymatique des xénobiotiques (substances étrangères). Au niveau moléculaire, ils peuvent perturber la transcription ADN-ARN ou la liaison à des récepteurs spécifiques cytoplasmiques et nucléaires, ou encore causer des lésions au niveau des gènes ou de leurs produits. Finalement, le dysfonctionnement d’un organe essentiel a de fortes chances d’être la conséquence d’une lésion moléculaire dans une cellule cible particulière de cet organe. Il n’est toutefois pas toujours faisable, ni nécessaire, de remonter jusqu’au mécanisme moléculaire: il est possible d’intervenir et de traiter sans connaître complètement la cible moléculaire. Néanmoins, la connaissance du mécanisme spécifique de la toxicité accroît la valeur prédictive et la pertinence d’une extrapolation à d’autres produits chimiques. La figure 33.8 représente de façon schématique les divers niveaux auxquels on peut détecter une interférence avec des processus physiologiques normaux. Les flèches montrant les conséquences chez un individu peuvent être interprétées dans un sens descendant (exposition, pharmacocinétique vers toxicité systémique ou organique) ou ascendant (lésion moléculaire, effet cellulaire ou biochimique vers toxicité systémique ou organique).
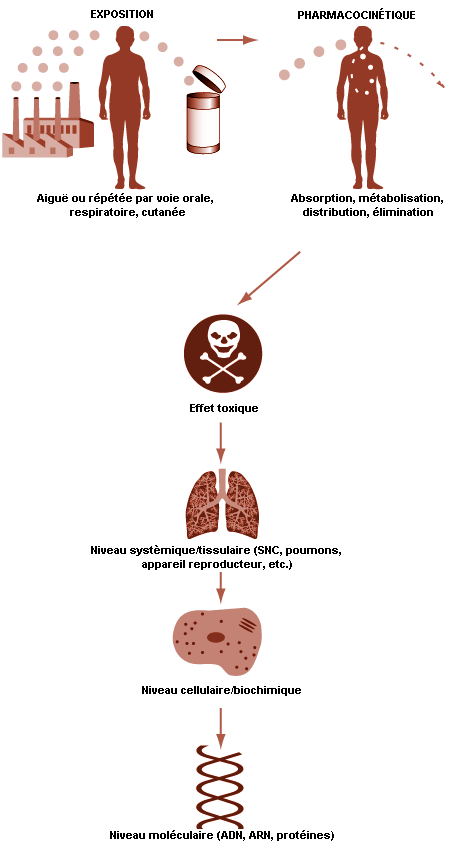
Les mécanismes de toxicité peuvent être simples ou complexes. On observe souvent une différence entre le type de toxicité, son mécanisme et le niveau de l’effet, selon que les effets nocifs sont dus à une seule exposition à dose élevée (cas d’une intoxication accidentelle), ou à une exposition répétée à plus faible dose (cas d’une exposition professionnelle ou environnementale). Traditionnellement, à des fins expérimentales, on administre soit une seule dose élevée ou dose aiguë par intubation directe dans l’estomac d’un rongeur, soit un gaz ou un produit volatil pendant deux à quatre heures par voie respiratoire, en choisissant la méthode qui reproduit le mieux les conditions d’une exposition humaine. Les animaux sont observés pendant une période de deux semaines après l’exposition, puis on procède à l’examen des principaux organes externes et internes pour la mise en évidence des lésions. Les études à doses répétées durent de quelques mois à plusieurs années. Chez les rongeurs, on considère qu’un suivi de deux années est suffisant pour évaluer la toxicité et la cancérogénicité en cas d’exposition chronique (sur toute une vie), alors que chez les primates non humains, on considère que pour une exposition subchronique (inférieure à la durée de vie), deux années sont la norme pour évaluer la toxicité de doses répétées. Après exposition, on effectue un examen complet de tous les tissus, organes et fluides pour déterminer les effets toxiques.
Les exemples suivants constituent une bonne illustration des effets aigus que l’on peut observer après une dose élevée pouvant entraîner la mort ou une incapacité grave. Dans certains cas, les effets peuvent être transitoires et complètement réversibles après traitement. Ils sont fonction de la dose ou de la sévérité de l’exposition.
Asphyxiants simples. Le mécanisme de toxicité des gaz inertes et de quelques autres substances non réactives est dû à un manque d’oxygène (anoxie). Ces produits chimiques, entraînant une privation en oxygène au niveau du système nerveux central (SNC), sont appelés asphyxiants simples. Lorsqu’un sujet pénètre dans un espace clos contenant de l’azote et peu d’oxygène, il va immédiatement manquer d’oxygène cérébral, ce qui va conduire à son inconscience et à son décès s’il n’est pas soustrait sur-le-champ à l’exposition. Dans des cas extrêmes (absence quasiment totale d’oxygène), cet état d’inconscience peut survenir en quelques secondes; la survie dépend donc de l’évacuation rapide vers un environnement oxygéné. Le sujet peut survivre mais présenter des dommages irréversibles au niveau cérébral si son sauvetage est retardé. Ces dommages irréversibles sont dus à la mort des neurones qui ne peuvent se régénérer.
Asphyxiants chimiques. Le monoxyde de carbone (CO) entre en concurrence avec l’oxygène pour se lier à l’hémoglobine (dans les hématies circulantes). Il prive les tissus de l’oxygène nécessaire au métabolisme énergétique, ce qui entraîne une mort cellulaire. L’intervention thérapeutique consiste à supprimer la source de CO et à traiter par de l’oxygène. L’utilisation d’oxygène est basée sur le mécanisme d’action toxique du CO. Un autre asphyxiant chimique puissant, le cyanure, perturbe le métabolisme cellulaire et l’utilisation de l’oxygène en formant de l’énergie. Le traitement par du nitrite de sodium transforme l’hémoglobine des hématies en méthémoglobine. L’ion cyanure présente une plus forte affinité pour la méthémoglobine que pour sa cible cellulaire. Par conséquent, la méthémoglobine capte le cyanure et l’éloigne des cellules cibles. C’est là la base de la thérapie antidotique.
Les dépresseurs du système nerveux central (SNC). Un grand nombre de produits, les solvants par exemple, provoquent, sous leur forme initiale ou après transformation en intermédiaires réactifs, une toxicité aiguë caractérisée par l’apparition d’une sédation ou d’un état d’inconscience. On pense que ces états sont dus à l’interaction du solvant avec les membranes cellulaires du SNC qui nuit à leur pouvoir de transmission des signaux électriques et chimiques. Bien que la sédation puisse paraître comme une forme bénigne de toxicité (on se souviendra que les premiers anesthésiques ont été développés sur ce principe), «seule la dose fait le poison». Si on administre à un animal une dose suffisante par ingestion ou inhalation, il peut mourir d’un arrêt respiratoire. Lorsque l’administration d’un produit anesthésique n’entraîne pas le décès du sujet, ce type d’intoxication est généralement facilement réversible si le patient est soustrait à l’exposition, si le produit chimique est redistribué dans l’organisme ou s’il en est éliminé.
Effets cutanés. Les effets nocifs au niveau de la peau vont, selon la substance en cause, de l’irritation à la corrosion. Les solutions acides et basiques fortes sont corrosives pour les tissus vivants et provoquent des brûlures chimiques avec cicatrices résiduelles. La cicatrisation est due à la mort des cellules dermiques responsables de la régénération. De plus faibles concentrations peuvent ne provoquer qu’une irritation de la première couche cutanée.
La sensibilisation chimique est un autre mécanisme toxique spécifique qui peut toucher la peau. Supposons, par exemple, que du 2,4-dinitrochlorobenzène se lie à certaines protéines cutanées. Le système immunitaire va alors considérer cette association comme un produit étranger et activer des cellules spéciales pour l’éliminer en sécrétant des médiateurs (cytokines), provoquant l’apparition d’un exanthème ou d’une dermatite (voir l’article «L’immunotoxicologie»). On assiste au même type de réaction du système immunitaire en présence du sumac vénéneux. La sensibilisation immunitaire est très spécifique d’un produit chimique donné et n’intervient qu’après deux expositions au moins. La première exposition sensibilise (reconnaissance du produit chimique par les cellules), les expositions suivantes déclenchant la réponse du système immunitaire. La suppression du contact et un traitement symptomatique avec des crèmes anti-inflammatoires contenant des stéroïdes suffisent généralement pour traiter les individus sensibilisés. Dans les cas sérieux ou réfractaires, on prescrit un immunosuppresseur systémique comme la prednisone ainsi qu’un traitement topique.
Sensibilisation pulmonaire. Le diisocyanate de toluène (TDI) provoque une réaction de sensibilisation immunitaire dont la cible est le poumon. Une surexposition au TDI chez des individus sensibles produit un œdème pulmonaire (accumulation liquidienne), une constriction bronchique et une gêne respiratoire. Ces cas, sérieux, nécessitent l’éviction du sujet de toute exposition potentielle ultérieure. Le traitement est essentiellement symptomatique. La sensibilisation cutanée et pulmonaire obéit à la relation dose-réponse. En cas de dépassement du niveau admis pour une exposition professionnelle, des effets nocifs peuvent apparaître.
Effets oculaires. L’atteinte de l’œil va de la simple rougeur de la couche externe (rougeur due à l’eau de piscine par exemple) à la lésion de la cornée ou de l’iris. Des tests d’irritation oculaire peuvent être conduits chez les animaux lorsqu’on pense qu’ils n’induiront pas de lésion sérieuse. De nombreux mécanismes provoquant une corrosion cutanée peuvent également être à l’origine d’une atteinte oculaire. Les substances corrosives pour la peau, comme les acides forts (pH inférieur à 2) et les bases fortes (pH supérieur à 11,5), ne sont pas testées sur les yeux des animaux, car elles entraîneraient une corrosion et la cécité par un mécanisme semblable à celui qui provoque la corrosion cutanée. Les agents tensioactifs comme les détergents et les surfactants peuvent quant à eux provoquer une atteinte de l’œil allant de l’irritation à la corrosion. Un groupe de substances à utiliser avec précaution est constitué des surfactants chargés positivement (cationiques), qui peuvent provoquer des brûlures, une opacité permanente de la cornée et une vascularisation (formation de vaisseaux sanguins). Un autre produit chimique, le dinitrophénol, est connu pour causer la formation de cataracte; celle-ci paraît être reliée à une concentration du produit au niveau de l’œil, ce qui constitue un exemple de distribution pharmacocinétique spécifique.
Bien que la liste ci-dessus soit loin d’être exhaustive, elle doit permettre au lecteur d’avoir un aperçu des divers mécanismes possibles de toxicité aiguë.
Certains produits chimiques ont un mécanisme de toxicité différent selon qu’ils sont administrés à dose unitaire élevée ou de manière répétée à dose plus faible quoique toxique. Lorsqu’on donne une forte dose unitaire, le sujet peut ne plus être en mesure de détoxifier et d’excréter le produit chimique et réagir différemment que si on lui administrait des doses répétées plus faibles. Le cas de l’alcool illustre le propos: de fortes doses d’alcool donnent lieu à des effets, principalement au niveau du système nerveux central, alors que des doses répétées plus faibles provoquent des lésions hépatiques.
Inhibition des anticholinestérasiques. La plupart des pesticides organophosphorés, par exemple, ont une faible toxicité chez les mammifères tant qu’ils ne sont pas activés par voie métabolique, essentiellement au niveau hépatique. Les organophosphorés agissent principalement en inhibant l’acétylcholinestérase (AChE) dans le cerveau et le système nerveux périphérique. L’AChE est l’enzyme normale qui inactive le neurotransmetteur acétylcholine. Une faible inhibition de l’AChE sur une période prolongée n’entraîne pas d’effets nocifs. Par contre, lorsque le niveau d’exposition est important, l’acétylcholine en excès cause une stimulation excessive du système nerveux cholinergique du fait de la forte inactivation de l’AChE. Celle-ci s’assortit de nombreux symptômes, dont l’arrêt respiratoire, qui peut évoluer vers le décès en cas d’absence de traitement. Pour y remédier, on administre principalement de l’atropine, qui bloque les effets de l’acétylcholine, et du chlorure de pralidoxime, qui réactive l’AChE inhibée. On voit à la lumière de cet exemple que la connaissance du mécanisme d’action biochimique des organophosphorés permet à la fois de comprendre le mécanisme de leur toxicité et de proposer un traitement spécifique.
Activation métabolique. De nombreux produits chimiques, dont le tétrachlorure de carbone, le chloroforme, l’acétylaminofluorène, les nitrosamines et le paraquat, sont activés par voie métabolique en radicaux libres ou intermédiaires réactifs inhibant ou perturbant le fonctionnement normal de la cellule. La mort cellulaire survient dans les expositions à fortes concentrations (voir l’article «La lésion et la mort cellulaires»). Bien que l’on ne connaisse pas encore les interactions et les cibles cellulaires spécifiques, tous les organes où a lieu l’activation de ces toxiques (foie, reins et poumons) constituent des cibles potentielles. Plus précisément, des cellules spécifiques à l’intérieur de ces organes sont douées de la propriété d’activer ou de détoxifier ces intermédiaires de façon plus ou moins efficace, propriété dont va dépendre la sensibilité intracellulaire de l’organe. Il est donc important de connaître la pharmacocinétique de ces produits, particulièrement les voies métaboliques, si l’on veut comprendre leur mécanisme d’action.
Mécanismes du cancer. Le cancer est une pathologie très hétérogène, et, bien que l’on commence à comprendre certains de ses mécanismes grâce aux techniques de biologie moléculaire mises au point depuis 1980, il reste encore beaucoup à apprendre. On sait de façon certaine que le cancer se développe selon un processus multiétapes et que des gènes critiques sont à l’origine de différents types de cancer. Des lésions de l’ADN (mutations somatiques) au niveau de ces gènes critiques peuvent être responsables d’une sensibilité accrue ou de l’apparition de lésions cancéreuses (voir l’article «La toxicologie génétique»). L’exposition à divers produits chimiques d’origine naturelle (par exemple, viande ou poisson cuisinés) ou synthétique (telle que la benzidine utilisée comme colorant), ou à des agents physiques (rayonnements ultraviolets, radon du sol, rayonnements gamma d’origine médicale ou industrielle) contribue à la formation de mutations géniques somatiques. Cependant, il existe des substances naturelles ou synthétiques (les antioxydants) et des processus de réparation de l’ADN qui ont un rôle protecteur et assurent l’homéostasie. Il est clair que la génétique est un facteur important dans le cancer, ainsi que le démontrent certaines maladies génétiques comme le xeroderma pigmentosum, où l’absence de mécanisme de réparation de l’ADN accroît sensiblement le risque de cancer cutané après une exposition aux rayonnements ultraviolets.
Mécanismes au niveau des organes reproducteurs. Comme pour le cancer, on sait qu’il existe de nombreux mécanismes de toxicité touchant la reproduction ou le développement, mais ces mécanismes sont encore mal élucidés. On sait que des virus (par exemple, la rubéole), certaines infections bactériennes ou des médicaments (comme le thalidomide, la vitamine A) ont un effet nocif sur le développement. Une étude de Khera (1991), rapportée par Carney (1994), a démontré chez l’animal que les anomalies du développement liées à l’éthylèneglycol sont dues à la formation de métabolites acides chez la mère. L’éthylèneglycol est métabolisé en métabolites acides, en particulier les acides glycolique et oxalique. Les effets sur le placenta et le fœtus paraissent en relation avec ce processus de toxification métabolique.
Notre propos, dans cet article, était d’examiner quelques mécanismes toxiques connus et de démontrer la nécessité de poursuivre ce type d’études. Il est à noter, cependant, que la connaissance de ces mécanismes n’est pas absolument nécessaire pour assurer la protection de la santé humaine et l’hygiène de l’environnement. Elle permet toutefois aux professionnels de mieux prévoir et gérer la toxicité. Les moyens à utiliser pour élucider les mécanismes de toxicité dépendent à la fois des connaissances de la communauté scientifique et de la réflexion des décideurs en matière de santé publique.
La science médicale dans son ensemble a pour vocation de prévenir la mort cellulaire, par exemple dans l’infarctus du myocarde, les traumatismes et les états de choc, ou de la provoquer, en particulier pour lutter contre les maladies infectieuses ou le cancer. Il est par conséquent essentiel de comprendre la nature des phénomènes qui entrent en jeu et leur mécanisme. On distingue deux types de mort cellulaire: la «mort accidentelle» due, par exemple, à des agents toxiques ou à une ischémie, et la «mort programmée» qui survient au cours du développement embryonnaire, comme lors de la formation des doigts ou de la résorption de la queue du têtard.
La lésion et la mort cellulaires sont donc des événements importants aussi bien en physiologie qu’en physiopathologie. La mort cellulaire physiologique est un phénomène capital pendant l’embryogenèse et le développement embryonnaire. L’étude de la mort cellulaire durant le développement a conduit à d’importants progrès dans la connaissance des mécanismes de génétique moléculaire, en particulier chez les invertébrés. Chez ces animaux, la localisation précise et le rôle des cellules entrant dans le processus de mort cellulaire ont fait l’objet de nombreuses études et, grâce à l’utilisation des techniques de mutagenèse, plusieurs gènes responsables ont pu être identifiés. Dans un organe adulte, l’équilibre entre mort cellulaire et prolifération cellulaire détermine la taille de l’organe. Dans certains organes, tels que la peau et l’intestin, les cellules se renouvellent en permanence. Pour prendre l’exemple de la peau, les cellules progressent vers la surface cutanée, où elles subissent une différenciation terminale et la mort cellulaire par kératinisation.
De nombreuses catégories de produits chimiques toxiques sont capables d’induire des lésions cellulaires aiguës entraînant la mort. On trouve, par exemple, des agents responsables d’anoxie et d’ischémie, tels que le cyanure de potassium; des cancérogènes chimiques, transformés en électrophiles se liant de manière covalente aux protéines des acides nucléiques; des produits chimiques oxydants, à l’origine de la formation de radicaux libres responsables de lésions oxydantes; des produits activant le complément; divers ionophores calciques. La mort cellulaire est également un volet important de la cancérogenèse chimique, de nombreux cancérogènes chimiques complets, à doses carcinogéniques, produisant une nécrose et une inflammation aiguës suivies d’une régénération et d’une prénéoplasie.
On entend par lésion cellulaire tout phénomène ou stimulus, par exemple un produit chimique, qui perturbe l’homéostasie de la cellule et permet ainsi la survenue d’un certain nombre d’événements. Les principales manifestations des lésions létales sont l’inhibition de la synthèse d’ATP, la rupture de l’intégrité de la membrane plasmique ou le déficit en facteurs de croissance essentiels (voir figure 33.9).
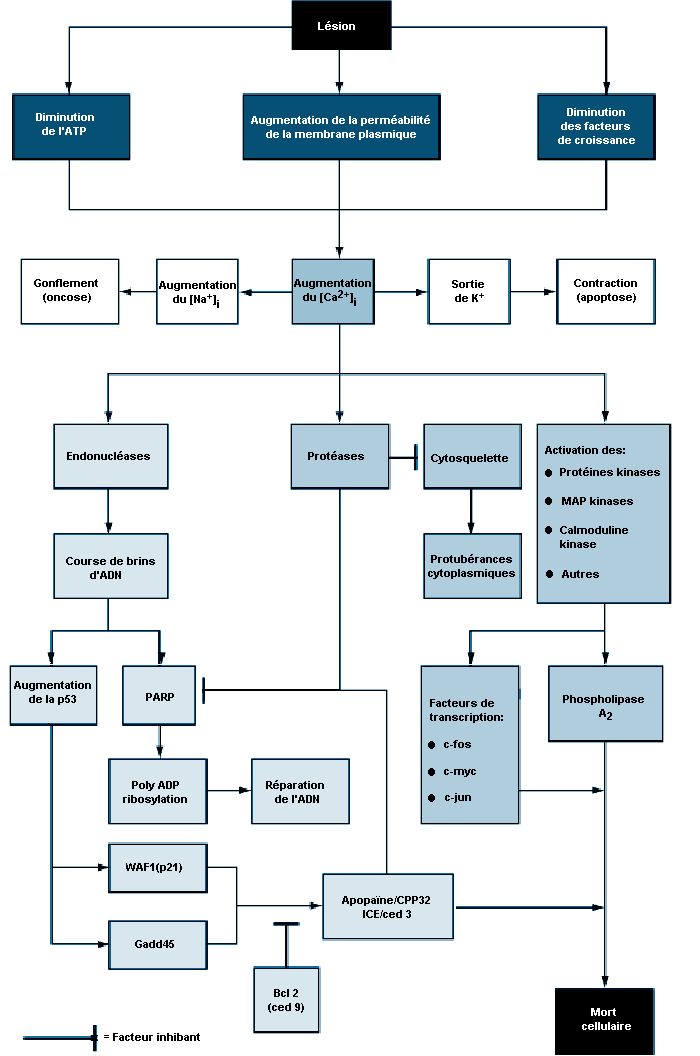
Les lésions létales entraînent la mort cellulaire après une période qui varie en fonction de la température, du type cellulaire et du stimulus; elles peuvent également être sublétales ou chroniques — la lésion n’aboutissant pas dans ce cas à la mort cellulaire, bien que l’homéostasie soit perturbée (Trump et Arstila, 1971; Trump et Berezesky, 1992; Trump et Berezesky, 1995; Trump, Berezesky et Osornio-Vargas, 1981). En cas de lésion létale, il se produit avant la mort cellulaire une phase appelée «phase prélétale». Si le stimulus lésionnel, une anoxie par exemple, disparaît au cours de cette période, la cellule peut récupérer; cependant, après un certain temps («point de non-retour» ou point de mort cellulaire), elle n’est plus en mesure de le faire, mais subit au contraire du fait de ce stimulus une dégradation et une hydrolyse. Ces processus se poursuivent jusqu’à ce qu’ils atteignent un état d’équilibre physico-chimique avec l’environnement; cette phase porte le nom de nécrose. Durant la phase prélétale, des modifications importantes se produisent qui varient en fonction du type de cellule et de lésion; il s’agit de l’apoptose et de l’oncose.
Le terme apoptose est dérivé du grec apo, signifiant «point éloigné», et ptosis, «chute». Il désigne le phénomène de condensation des cellules et de formation de protubérances à la périphérie qui survient à la faveur de ce changement prélétal. Ces protubérances se détachent ensuite et dérivent à l’état libre. L’apoptose se produit dans divers types de cellules à la suite de lésions toxiques diverses elles aussi (Wyllie, Kerr et Currie, 1980). Elle frappe particulièrement les lymphocytes, où elle constitue le mécanisme prédominant du renouvellement des clones lymphocytaires. Les fragments résultant du phénomène forment des corps basophiles à l’intérieur des macrophages des ganglions lymphatiques. Dans les autres organes, l’apoptose se produit dans des cellules isolées, les fragments formés sont rapidement éliminés par les cellules parenchymateuses adjacentes ou les macrophages, avant et après la mort par phagocytose. L’apoptose de cellules isolées suivie d’une phagocytose n’est pas associée à un état inflammatoire. Avant de mourir, les cellules apoptotiques ont un cytosol très dense avec des mitochondries normales ou condensées. Le réticulum endoplasmique est normal ou légèrement dilaté. La chromatine nucléaire est très nettement agglomérée le long de l’enveloppe nucléaire et autour du nucléole. Le contour nucléaire est également irrégulier et une fragmentation nucléaire se produit. La condensation de la chromatine s’accompagne d’une fragmentation de l’ADN qui, dans la plupart des cas, a lieu entre les nucléosomes, donnant un aspect caractéristique à l’électrophorèse en échelle.
Dans l’apoptose, une augmentation du [Ca2+]i stimule la sortie du K+ et produit une condensation cellulaire, mécanisme nécessitant probablement de l’ATP. Les lésions qui inhibent totalement la synthèse de l’ATP entraînent donc très vraisemblablement une apoptose. Une augmentation prolongée du [Ca2+]i est à l’origine de nombreux effets délétères, en particulier l’activation de protéases, d’endonucléases et de phospholipases. L’activation des endonucléases est responsable de coupures d’ADN simple ou double brin qui, à leur tour, provoquent une augmentation du taux de p53, de la poly-ADP ribosylation et des protéines nucléaires essentielles à la réparation de l’ADN. L’activation des protéases modifie un certain nombre de substrats, dont l’actine et les protéines associées, conduisant à la formation des protubérances. La poly(ADP-ribose) polymérase (PARP), autre substrat important, inhibe la réparation de l’ADN. L’augmentation du [Ca2+]i est également associée à l’activation d’un certain nombre de protéines kinases, telles que la MAP kinase, la calmoduline kinase, entre autres. Ces kinases participent à l’activation de facteurs de transcription initiant la transcription de gènes tels que c-fos, c-jun et c-myc et à l’activation de la phospholipase A2, qui provoque une augmentation de la perméabilité de la membrane plasmique et des membranes intracellulaires telles que la membrane mitochondriale interne.
Mot dérivé du grec onkos, qui signifie «masse, volume», l’oncose doit son nom au fait que, dans ce type de perturbation prélétale, la cellule commence par devenir plus volumineuse presque immédiatement après une lésion (Majno et Joris, 1995) en raison d’une augmentation de la concentration en cations dans le cytosol cellulaire. Le principal cation responsable de ce changement de volume est le sodium, normalement régulé pour maintenir le volume cellulaire. Cependant, en absence d’ATP ou si la Na-ATPase du plasmalemme est inhibée, les protéines intracellulaires cessent d’assurer le contrôle du volume et le sodium cytosolique augmente. Parmi les phénomènes précoces de l’oncose on trouve, par conséquent, l’augmentation du [Na+]i qui conduit au changement de volume cellulaire et celle du [Ca2+]i résultant soit d’un influx à partir de l’espace extracellulaire, soit de sa libération à partir des lieux de stockage intracellulaires. Il en résulte un gonflement du cytosol, du réticulum endoplasmique, de l’appareil de Golgi et la formation de vésicules aqueuses autour de la surface cellulaire. Les mitochondries subissent dans un premier temps une condensation, suivie ultérieurement d’un gonflement très important lié aux lésions de la membrane mitochondriale interne. Dans ce type de lésion prélétale, la chromatine finit par se dégrader après avoir subi une condensation; néanmoins, le profil en échelle caractéristique de l’apoptose n’apparaît pas.
La nécrose correspond à une série de modifications se produisant après la mort cellulaire lorsque la cellule est transformée en débris éliminés par la réponse inflammatoire. On distingue deux types de nécrose: la nécrose oncotique et la nécrose apoptotique. La nécrose oncotique se produit dans des zones étendues, lors d’un infarctus du myocarde, par exemple, ou localement dans un organe après une atteinte toxique de nature chimique, notamment au niveau du tubule rénal proximal après administration de HgCl2. Des zones étendues d’un organe sont touchées et les cellules nécrotiques induisent rapidement une réaction inflammatoire, d’abord aiguë puis chronique. Si l’organisme survit, on assiste dans de nombreux organes à l’élimination des cellules mortes et à une régénération; c’est notamment ce qui se passe dans le foie ou le rein à la suite d’une intoxication chimique. Au contraire, la nécrose apoptotique se produit d’ordinaire dans une cellule unique et les débris nécrotiques sont formés à l’intérieur des phagocytes des macrophages ou des cellules du parenchyme adjacent. A un stade très précoce, les cellules nécrotiques se caractérisent par une interruption de la continuité de la membrane plasmique et par des densités floconneuses provenant des protéines dénaturées de la matrice mitochondriale. Dans certaines formes de lésion n’interférant pas avec l’accumulation de calcium mitochondrial, des dépôts de phosphates de calcium peuvent être observés à l’intérieur des mitochondries. D’autres membranes sont elles aussi fragmentées: le RE, les lysosomes et l’appareil de Golgi. Finalement, la chromatine nucléaire subit une lyse, résultant de l’action des hydrolases lysosomiales. Après la mort cellulaire, les hydrolases lysosomiales jouent un rôle important dans l’élimination des débris par l’intermédiaire des cathepsines, nucléolases et lipases qui, en raison de leur pH acide optimal, peuvent survivre au faible pH des cellules nécrotiques, les autres enzymes cellulaires étant dénaturées et inactivées.
Lors des atteintes létales, les interactions initiales les plus habituelles conduisant à la mort cellulaire sont celles qui perturbent d’une part le métabolisme énergétique — telles que l’anoxie, l’ischémie ou les inhibiteurs de la respiration cellulaire — et, de l’autre, la glycolyse, comme c’est le cas en présence de cyanure de potassium, de monoxyde de carbone ou d’iodo-acétate, pour ne citer que ces produits. Nous l’avons déjà mentionné, les produits inhibant le métabolisme énergétique aboutissent à l’oncose s’ils sont présents en forte concentration. Il est un autre type habituel de lésion initiale évoluant rapidement vers la mort cellulaire: la modification des fonctions membranaires plasmiques (Trump et Arstila, 1971; Trump, Berezesky et Osornio-Vargas, 1981). Ce type de modification peut prendre la forme d’une lésion directe avec augmentation de la perméabilité membranaire, cas observé lors de traumatismes, d’une activation du complexe C5b-C9 du complément, d’une lésion mécanique de la membrane cellulaire ou de l’inhibition de la pompe sodium-potassium (Na+-K+) par des glucosides tels que l’ouabaïne. Les ionophores calciques comme l’ionomycine ou l’A23187, en provoquant une augmentation rapide de la concentration du [Ca2+] intracellulaire, sont également responsables d’une lésion létale aiguë. Selon les cas, le type de lésion prélétale est soit l’apoptose, soit l’oncose.
Dans de nombreux types de lésion, la respiration mitochondriale et la phosphorylation oxydative sont rapidement touchées. Dans certaines cellules, il en résulte une stimulation de la glycolyse anaérobie permettant de maintenir l’ATP, mais ce processus est inhibé lors de nombreuses atteintes. Un déficit en ATP prive de nombreux processus homéostatiques importants de l’énergie nécessaire, en particulier, le contrôle de l’homéostasie ionique intracellulaire (Trump et Berezesky, 1992; Trump, Berezesky et Osornio-Vargas, 1981). Ce déficit provoque une augmentation rapide du [Ca2+]i, du [Na+] et du [Cl-] responsables du gonflement cellulaire. L’élévation du [Ca2+]i entraîne l’activation d’un certain nombre d’autres mécanismes de signalisation traités ci-après, dont diverses kinases, pouvant provoquer une augmentation rapide de la transcription génique de proto-oncogènes. L’augmentation du [Ca2+]i modifie également le fonctionnement du cytosquelette, partiellement responsable de la formation de protubérances et de l’activation d’endonucléases, de protéases et de phospholipases. Celles-ci semblent déclencher un grand nombre des effets importants mentionnés précédemment: lésions membranaires par suite de l’activation des protéases et des lipases, dégradation directe de l’ADN par activation des endonucléases et activation des kinases telles que la MAP kinase et la calmoduline kinase, agissant comme facteurs de transcription.
Des études approfondies chez les invertébrés C. elegans et Drosophila et sur cellules humaines et animales ont permis d’identifier une série de gènes facteurs de mort. Certains de ces gènes d’invertébrés ont leurs homologues chez les mammifères. Par exemple, le gène ced-3, essentiel à la mort cellulaire programmée chez C. elegans, possède une activité protéasique et présente une forte homologie avec l’enzyme de conversion de l’interleukine des mammifères. Un gène très proche appelé apopaïne ou prICE a récemment été identifié avec une homologie encore plus étroite (Nicholson et coll., 1995). Chez la Drosophila, le gène facteur de mort semble être impliqué dans un signal conduisant à la mort cellulaire programmée. Les autres gènes facteurs de mort comprennent celui de la protéine membranaire Fas et l’important gène suppresseur de tumeurs qu’est le p53. La protéine p53 est induite à la suite de lésions sur l’ADN et, une fois phosphorylée, elle agit comme facteur de transcription pour d’autres gènes tels que gadd45 et waf-1, impliqués dans la signalisation de la mort cellulaire. D’autres proto-oncogènes comme c-fos, c-jun et c-myc semblent également intervenir dans certains systèmes.
De façon parallèle, il existe des gènes antimort qui semblent contrecarrer les gènes facteurs de mort. Le premier d’entre eux à avoir été identifié est le ced-9 chez C. elegans, homologue du bcl-2 chez l’humain. Ces gènes agissent de manière encore inconnue pour empêcher la mort cellulaire due soit à des mécanismes génétiques, soit à des produits chimiques toxiques. De récentes découvertes montrent que le bcl-2 agirait comme antioxydant. Actuellement, des efforts importants sont déployés pour mieux comprendre les gènes qui interviennent dans ces phénomènes et pour trouver les moyens de les activer ou de les inhiber selon le cas.
La toxicologie génétique est l’étude du rôle des agents chimiques ou physiques dans le processus complexe de l’hérédité. Les produits chimiques génotoxiques sont les composés capables de modifier le matériel héréditaire des cellules vivantes. La probabilité pour qu’un produit chimique provoque une lésion génétique dépend nécessairement de plusieurs variables, en particulier le taux d’exposition de l’organisme au produit chimique, sa distribution et sa rétention une fois qu’il est entré dans l’organisme, l’efficacité de l’activation métabolique ou des systèmes de détoxification au niveau des cibles tissulaires et la réactivité du produit chimique ou de ses métabolites avec les macromolécules critiques à l’intérieur des cellules. La probabilité qu’une lésion génétique provoque une maladie dépend de plusieurs facteurs: nature de la lésion, capacité de la cellule de réparer ou d’amplifier la lésion génétique, possibilité pour une lésion de s’exprimer et aptitude de l’organisme à reconnaître et à bloquer la multiplication des cellules aberrantes.
Dans les organismes supérieurs, l’information héréditaire est organisée au niveau des chromosomes. Ceux-ci sont constitués de brins fortement condensés d’ADN en association avec des protéines. Dans un chromosome, chaque molécule d’ADN est constituée d’une paire de longues chaînes, non ramifiées, de sous-unités nucléotidiques associées par des liens phosphodiester joignant le carbone 5 d’un désoxyribose au carbone 3 du désoxyribose suivant (voir figure 33.10). De plus, l’une des quatre bases nucléotidiques (adénine, cytosine, guanine ou thymine) est attachée à chaque sous-unité désoxyribose à l’instar des perles d’un collier. Du point de vue tridimensionnel, chaque paire de brins d’ADN forme une double hélice avec l’ensemble des bases orientées vers l’intérieur de la spirale. A l’intérieur de l’hélice, chaque base est associée à sa base complémentaire sur le brin d’ADN opposé; une liaison hydrogène impose un appariement solide, non covalent, de l’adénine avec la thymine et de la guanine avec la cytosine (voir figure 33.10). Etant donné que la séquence des bases nucléotidiques est complémentaire sur toute la longueur de la double molécule d’ADN, les deux brins portent fondamentalement la même information génétique. En fait, lors de la réplication de l’ADN, chaque brin sert de matrice pour produire un nouveau brin fils.
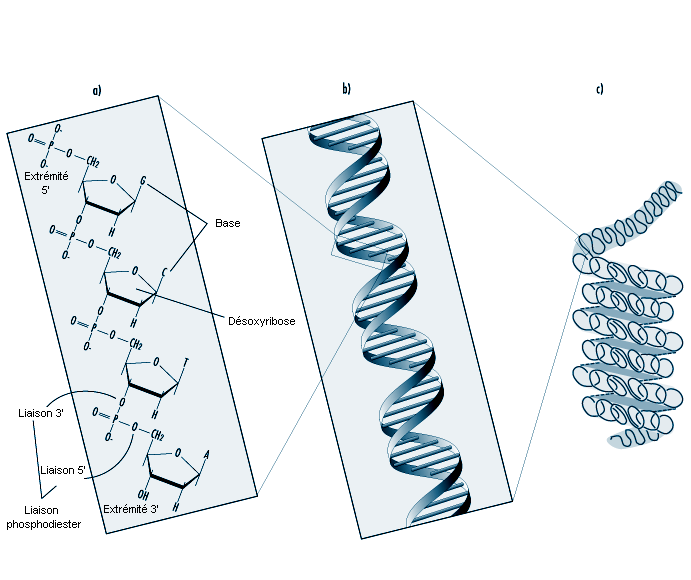
A l’aide de l’ARN et d’une série de protéines, la cellule transcrit finalement l’information codée par la séquence linéaire des bases au niveau de régions spécifiques de l’ADN (gènes) et produit les protéines essentielles à la survie cellulaire ainsi qu’à la croissance ou à la différenciation normales. On peut donc dire que les nucléotides font office d’alphabet biologique servant au codage des acides aminés, unités chimiques de base des protéines.
Lorsqu’il se produit une perte de nucléotides ou une insertion de nucléotides incorrects, ou encore lorsque des nucléotides inutiles sont ajoutés lors de la synthèse de l’ADN, l’erreur est appelée une mutation. On estime qu’il se produit moins d’une mutation tous les 109 nucléotides incorporés lors de la réplication cellulaire normale. Bien que les mutations ne soient pas nécessairement dangereuses, les altérations provoquant l’inactivation ou la surexpression de gènes importants peuvent aboutir à des troubles divers, dont le cancer, à des maladies héréditaires, des anomalies du développement, une stérilité ou la mort embryonnaire ou périnatale. Il est rare qu’une mutation entraîne un prolongement de la vie; ces événements forment la base de la sélection naturelle.
Bien que certains produits chimiques réagissent directement avec l’ADN, la plupart ont besoin pour cela d’une activation métabolique. Dans ce cas, des intermédiaires électrophiles tels que les époxydes ou les ions carbonium sont les responsables ultimes des lésions induites sur divers sites nucléophiles du matériel génétique (voir figure 33.11). En d’autres circonstances, la génotoxicité est due à des sous-produits de l’interaction d’une substance avec les lipides, les protéines intracellulaires ou l’oxygène.
En raison de leur relative abondance dans les cellules, les protéines sont la cible la plus fréquente d’une interaction toxique. Cependant, la modification de l’ADN est plus inquiétante étant donné le rôle central que joue cette molécule dans la régulation de la croissance et de la différenciation tout au long des générations cellulaires successives.
Au niveau moléculaire, les composés électrophiles attaquent les atomes d’oxygène et d’azote sur l’ADN. La figure 33.12 montre les sites les plus susceptibles de subir de telles modifications. Bien que les atomes d’oxygène des groupements phosphates de la chaîne principale de l’ADN soient également la cible des modifications chimiques, on estime que les lésions des bases sont plus intéressantes du point de vue biologique, car elles sont des éléments d’information essentiels de la molécule d’ADN.
Les composés comportant une partie électrophile ont un pouvoir génotoxique du fait de la formation de mono-adduits sur l’ADN. De la même façon, les composés ayant deux ou plusieurs parties réactives peuvent réagir avec deux centres nucléophiles différents et entraîner ainsi la formation de ponts intra- ou intermoléculaires dans le matériel génétique (voir figure 33.13). Les pontages interbrins ADN-ADN et les pontages ADN-protéine sont particulièrement cytotoxiques puisqu’ils peuvent causer un blocage total de la réplication de l’ADN. Pour des raisons évidentes, une cellule morte ne peut pas subir de mutation ou devenir néoplasique. Les agents génotoxiques peuvent aussi provoquer des cassures de l’épine dorsale phosphodiester, ou entre les bases et les sucres (produisant des sites apuriniques ou apyrimidiques) sur l’ADN. De telles cassures peuvent être le résultat direct de la réactivité chimique sur un site lésionnel, ou se produire lors de la réparation d’un des types de lésions de l’ADN mentionnés ci-dessus.
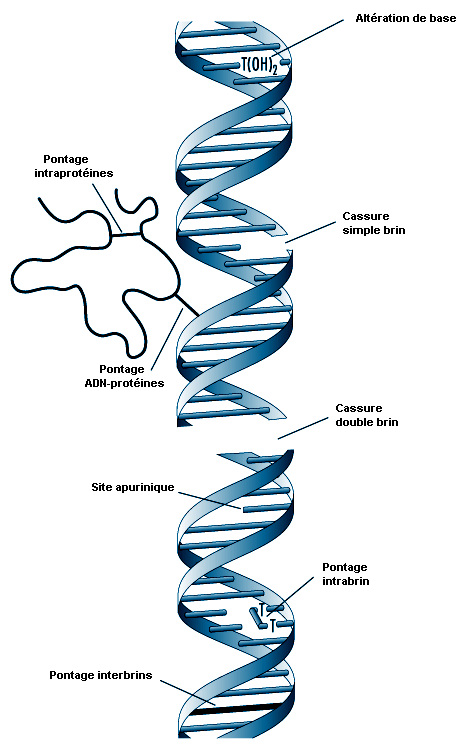
Les trente à quarante dernières années ont été marquées par la mise au point de toute une série de techniques pour surveiller les dommages génétiques induits par divers produits chimiques. Ces essais sont détaillés dans une autre section du présent chapitre, ainsi que dans d’autres chapitres de l’Encyclopédie.
Les erreurs de réplication des «microlésions» telles que les mono-adduits, les sites apuriniques ou apyrimidiques ou les cassures simple brin peuvent conduire finalement à des substitutions de paires de bases nucléotidiques, ou à l’insertion ou à la délétion de fragments polynucléotidiques courts dans l’ADN chromosomique. Au contraire, les «macrolésions», telles que les adduits volumineux, les pontages ou les cassures double brin, peuvent déclencher l’acquisition, la perte ou le réarrangement de fragments relativement importants de chromosomes. Quoi qu’il en soit, les conséquences peuvent être dévastatrices pour l’organisme, chacun de ces événements pouvant conduire à la mort cellulaire, à la perte d’une fonction ou à une transformation cellulaire maligne. On ne sait pratiquement rien de la façon dont les lésions de l’ADN induisent un cancer. On pense actuellement qu’il se produit une activation inappropriée de proto-oncogènes tels que myc et ras, ou l’inactivation de gènes suppresseurs de tumeur récemment identifiés tels que le p53. L’expression anormale de l’un de ces gènes détruit les mécanismes cellulaires normaux assurant le contrôle de la prolifération ou de la différenciation cellulaires.
Les études expérimentales montrent pour la plupart que le développement d’un cancer après une exposition à des composés électrophiles est un événement relativement rare. Une des explications en est que la cellule est douée de la capacité intrinsèque de reconnaître et de réparer l’ADN endommagé ou que les cellules dont l’ADN est endommagé ne sont pas capables de survivre. Durant la réparation, la base endommagée, le nucléotide ou la courte séquence de nucléotides autour du site lésé sont supprimés et, grâce au brin opposé qui sert de matrice, une nouvelle séquence d’ADN est synthétisée et mise en place. Pour être efficace, la réparation de l’ADN doit s’effectuer avant la division cellulaire et avoir une grande précision, pour éviter toute possibilité de propager une mutation.
Des études cliniques ont montré que les sujets présentant des déficits héréditaires des mécanismes de réparation de l’ADN manifestent fréquemment un cancer ou des anomalies du développement à un âge précoce (voir tableau 33.4). De tels exemples soulignent le lien évident entre les lésions de l’ADN et la pathologie humaine. De la même façon, des agents favorisant la prolifération cellulaire (tels que l’acétate de tétradécanoylphorbol) intensifient souvent la cancérogenèse. Pour ces composés, la probabilité élevée de transformation néoplasique est sans doute la conséquence du temps plus court dont dispose la cellule pour effectuer une réparation correcte de son ADN.
|
Syndrome |
Symptômes |
Phénotype cellulaire |
|
Ataxie télangiectasique |
Atteinte neurologique |
Hypersensibilité aux rayonnements ionisants et à certains agents alkylants |
|
Syndrome de Bloom |
Anomalies du développement |
Fréquence élevée d�aberrations chromosomiques |
|
Anémie de Fanconi |
Retard de croissance |
Hypersensibilité aux agents intercalants |
|
Cancer du côlon héréditaire non polyposique |
Forte incidence de cancer du côlon |
Défaut de réparation des erreurs d�appariement au niveau de l�ADN (erreur d�insertion de nucléotide lors de la réplication) |
|
Xeroderma pigmentosum |
Forte incidence d�épithélioma sur les zones cutanées exposées |
Hypersensibilité à la lumière UV et à de nombreux cancérogènes chimiques |
Les théories les plus anciennes sur l’interaction des produits chimiques avec l’ADN datent des études conduites lors de la mise au point du gaz moutarde comme gaz de combat. Par la suite, les efforts déployés pour identifier les agents anticancéreux susceptibles de bloquer sélectivement la réplication des cellules tumorales ont peu à peu permis de mieux comprendre cette interaction. L’intérêt accru du public pour les risques environnementaux a incité à poursuivre les recherches sur les mécanismes et les conséquences de l’interaction d’un produit chimique avec le matériel génétique. Le tableau 33.5 recense certains des produits chimiques ayant un pouvoir génotoxique.
|
Classe du produit chimique |
Exemple |
Origine de l�exposition |
Lésion génotoxique probable |
|
Aflatoxines |
Aflatoxine B1 |
Aliments contaminés |
Adduits de grande taille sur l�ADN |
|
Amines aromatiques |
2-Acétylaminofluorène |
Environnement |
Adduits de grande taille sur l�ADN |
|
Quinones aziridine |
Mitomycine C |
Chimiothérapie anticancéreuse |
Mono-adduits, pontages interbrins et cassures simple brin dans l�ADN |
|
Hydrocarbures chlorés |
Chlorure de vinyle |
Environnement |
Mono-adduits de l�ADN |
|
Métaux et composés métalliques |
Cisplatine |
Chimiothérapie anticancéreuse |
Pontages intrabrin et interbrins dans l�ADN |
|
|
Composés du nickel |
Environnement |
Mono-adduits et cassures simple brin dans l�ADN |
|
Moutardes azotées |
Cyclophosphamide |
Chimiothérapie anticancéreuse |
Mono-adduits et pontages interbrins dans l�ADN |
|
Nitrosamines |
N-Nitrosodiméthylamine |
Aliments contaminés |
Mono-adduits de l�ADN |
|
Hydrocarbures aromatiques polycycliques |
Benzo(a)pyrène |
Environnement |
Adduits de grande taille sur l�ADN |
Le système immunitaire a pour fonction de protéger l’organisme contre les agents infectieux qui peuvent l’envahir et d’assurer une surveillance immunologique vis-à-vis des cellules tumorales naissantes. Il constitue une première ligne de défense non spécifique capable d’amorcer des réactions effectrices et une voie spécifique acquise dans laquelle les lymphocytes et les anticorps possèdent une spécificité leur permettant de reconnaître l’antigène puis d’y réagir.
L’immunotoxicologie est la discipline qui étudie les événements conduisant à des effets nocifs dus à l’interaction des xénobiotiques avec le système immunitaire. Ces événements indésirables peuvent résulter: 1) d’un effet direct ou indirect du xénobiotique (ou de son produit de biotransformation) sur le système immunitaire; 2) d’une réponse immunologique de l’hôte au composé ou à son (ses) métabolite(s), ou aux antigènes de l’hôte modifiés par le composé ou ses métabolites (Berlin et coll., 1987).
Lorsque le système immunitaire agit comme cible passive des agressions chimiques, il peut offrir une résistance moindre aux infections et à certaines formes de néoplasie, ou être le siège d’un dérèglement ou d’une stimulation immunitaire pouvant aggraver une allergie ou une auto-immunité. Lorsqu’il répond à la spécificité antigénique du xénobiotique ou à l’antigène de l’hôte modifié par le toxique, la toxicité s’exprime par des allergies ou des maladies auto-immunes.
Des modèles animaux, dont certains sont validés, ont été mis au point pour étudier l’immunosuppression induite par un produit chimique (Burleson, Munson et Dean, 1995; PISSC, 1996). Lors d’une évaluation, on procède par étapes pour faire une sélection parmi les très nombreux tests disponibles. Généralement, lors de la première étape, on identifie les immunotoxiques potentiels. En cas de réponse positive, on passe à l’étape suivante qui consiste à confirmer et à mieux caractériser les perturbations observées. La troisième étape porte spécifiquement sur le mécanisme d’action du produit. Des études de ce type effectuées sur des animaux de laboratoire ont permis de constater que plusieurs xénobiotiques agissent comme immunosuppresseurs.
Les informations dont on dispose sur les perturbations de la fonction immunitaire chez l’humain par des produits chimiques environnementaux ne sont pas très abondantes (Descotes, 1986; NRC, 1992). L’utilisation d’indicateurs d’immunotoxicité n’est pas très répandue dans les études cliniques et épidémiologiques concernant l’effet des produits chimiques sur la santé. De telles études ne sont pas fréquentes, et quand bien même on en effectue, leur interprétation ne permet pas toujours de tirer des conclusions sans équivoque, notamment parce qu’il s’avère impossible de contrôler les expositions. Par conséquent, jusqu’à présent, les décisions concernant le danger et l’estimation du risque sont fondées sur des évaluations de l’immunotoxicité chez les rongeurs, avec extrapolation à l’humain.
Les réactions d’hypersensibilité, notamment l’asthme allergique et l’eczéma de contact, sont des problèmes de santé professionnelle importants dans les pays industriels (Vos, Younes et Smith, 1995). Le phénomène de sensibilisation de contact a été étudié en premier chez le cobaye (Andersen et Maibach, 1985); jusqu’à récemment, c’était l’espèce de choix pour les tests prédictifs. De nombreux tests sur cet animal sont disponibles, les plus fréquemment employés étant le test de maximisation et le patch test de Buehler. Les tests sur cobaye et des méthodes plus récentes mises au point chez la souris, comme le test de l’œdème de l’oreille ou celui des ganglions lymphatiques locaux, fournissent au toxicologue les outils pour évaluer le risque de sensibilisation cutanée. S’agissant de la sensibilisation au niveau de l’appareil respiratoire, la situation est très différente. Actuellement, il n’existe pas encore de méthode bien validée ou largement acceptée pour identifier les produits chimiques allergènes à ce niveau, bien que des progrès aient été accomplis sur des modèles animaux faisant appel au cobaye et à la souris.
Des données recueillies chez l’être humain montrent que des agents chimiques, des médicaments en particulier, peuvent provoquer des maladies auto-immunes (Kammüller, Bloksma et Seinen, 1989). Il existe un certain nombre de modèles animaux de maladies auto-immunes humaines, notamment pour des pathologies spontanées (comme le lupus érythémateux systémique chez la souris New Zealand Black), ou des phénomènes auto-immuns induits par immunisation expérimentale avec un auto-antigène réagissant de façon croisée (arthrite induite par l’adjuvant H37Ra chez le rat Lewis). Ces modèles sont utilisés pour l’évaluation préclinique de médicaments immunosuppresseurs. Très peu d’études ont fait appel à ces modèles pour établir si un xénobiotique potentialise l’auto-immunité induite ou congénitale. En réalité, les modèles animaux dont on peut se servir pour étudier la capacité des produits chimiques d’induire des maladies auto-immunes sont très peu nombreux. Un modèle employé de façon limitée est le test du ganglion poplité chez la souris. Comme c’est le cas chez l’être humain, les facteurs génétiques jouent un rôle capital dans le développement des maladies auto-immunes chez les animaux de laboratoire, ce qui limite la valeur prédictive de ces tests.
Le système immunitaire a pour principale fonction d’assurer la défense contre les bactéries, virus, parasites, champignons et cellules néoplasiques. Cette défense est confiée à divers types cellulaires et leurs médiateurs solubles dans une harmonie finement réglée. La défense de l’hôte peut être grossièrement divisée en résistance non spécifique ou innée et en immunité spécifique ou acquise sous la dépendance des lymphocytes (Roitt, Brostoff et Male, 1989).
Les composants du système immunitaire sont présents dans l’ensemble de l’organisme (Jones et coll., 1990). Le compartiment lymphocytaire se retrouve au niveau des organes lymphoïdes (voir figure 33.14). La moelle osseuse et le thymus sont classés comme organes lymphoïdes primaires ou centraux; les organes lymphoïdes secondaires ou périphériques comprennent les ganglions lymphatiques, la rate et le tissu lymphoïde situé le long de surfaces sécrétoires telles que les tractus respiratoire et gastro-intestinal, encore appelé tissu lymphoïde associé aux muqueuses (TLAM). Environ la moitié des lymphocytes de l’organisme sont localisés dans le TLAM, quel que soit le moment considéré. De plus, la peau est un organe important pour l’induction de réponses immunitaires contre les antigènes présents à son niveau. Les cellules épidermiques de Langerhans jouent un rôle important dans ce processus du fait de leur rôle dans la présentation de l’antigène.
Les cellules phagocytaires de la lignée monocyte/macrophage, ou système phagocytaire mononucléé (SPM), se trouvent dans les organes lymphoïdes ainsi que dans des sites extraganglionnaires; les phagocytes extraganglionnaires comprennent les cellules de Kupffer du foie, les macrophages alvéolaires au niveau pulmonaire et les macrophages mésangiaux respectivement aux niveau pulmonaire et rénal et les cellules gliales dans le cerveau. Les leucocytes polynucléaires sont localisés principalement dans le sang et la moelle osseuse, mais s’accumulent aux sites d’inflammation.
Une première ligne de défense contre les micro-organismes est assurée par une barrière physique et chimique: la peau et le tractus respiratoire ou digestif. Cette barrière est secondée par des mécanismes de protection non spécifiques comprenant les cellules phagocytaires (macrophages et leucocytes polynucléaires), capables de tuer les agents pathogènes, et les cellules tueuses naturelles, qui peuvent lyser les cellules tumorales et les cellules infectées par des virus. Le système du complément et certains inhibiteurs microbiens (par exemple, le lysozyme) prennent également part à la réponse immunitaire.
Après un premier contact entre l’hôte et un agent pathogène, on assiste à une série de réponses immunitaires spécifiques. Ce qui caractérise cette seconde ligne de défense c’est qu’elle est capable de distinguer les déterminants antigéniques, ou épitopes, des agents pathogènes grâce à des récepteurs présents à la surface des lymphocytes B et T. Après interaction avec les antigènes spécifiques, la cellule portant ce récepteur est stimulée, ce qui provoque sa prolifération et sa différenciation et aboutit à un clone de cellules filles propres à l’antigène responsable. Les réponses immunitaires spécifiques complètent la défense non spécifique opposée aux agents pathogènes en stimulant l’efficacité des réponses non spécifiques. L’immunité spécifique a pour caractéristique fondamentale de créer une mémoire. Une seconde exposition au même antigène provoque une réponse mieux régulée, plus rapide et plus intense.
Le génome n’est pas à même de contenir les codes de l’ensemble des récepteurs antigéniques dont il faut disposer pour reconnaître les nombreux antigènes pouvant être rencontrés. Le répertoire de spécificité se développe selon un processus de réarrangement génique qui est le pur fruit du hasard. Diverses spécificités apparaissent, dont certaines sont indésirables lorsqu’elles sont dirigées contre les composants du soi. Un processus de sélection au niveau du thymus (cellules T) ou de la moelle osseuse (cellules B) intervient pour éliminer ces spécificités indésirables.
La fonction effectrice immunitaire et la régulation homéostasique de la réponse immunitaire dépendent d’une série de produits solubles, les cytokines. Synthétisées et sécrétées par les lymphocytes et d’autres types cellulaires, les cytokines ont des effets pléiotropes sur les réponses immunitaires et inflammatoires. La réponse immunitaire ne peut se faire sans une coopération entre les différentes populations cellulaires. Cette coopération prend différentes formes: régulation des réponses des anticorps, afflux des cellules et des molécules immunitaires aux sites inflammatoires, initiation des réponses en phase aiguë, contrôle de la fonction cytotoxique des macrophages et nombreux autres processus essentiels à la résistance de l’hôte. Ces processus sont influencés par les cytokines agissant individuellement ou en association et, bien souvent, en dépendent.
L’immunité spécifique comporte deux voies: l’immunité à médiation humorale et l’immunité à médiation cellulaire.
Immunité à médiation humorale. Dans cette voie humorale, les lymphocytes B sont stimulés après la reconnaissance de l’antigène par les récepteurs de surface. Les récepteurs antigéniques sur les lymphocytes B sont des immunoglobulines (Ig). Les cellules B matures (cellules plasmatiques) commencent à produire des immunoglobulines spécifiques de l’antigène, qui agissent comme anticorps au niveau sérique et au niveau des surfaces muqueuses. On distingue cinq catégories principales d’immunoglobulines: 1) l’IgM, immunoglobuline pentamère, à capacité optimale d’agglutination, la première à être produite après une stimulation antigénique; 2) l’IgG, principale immunoglobuline dans la circulation, qui peut passer dans le placenta; 3) l’IgA, immunoglobuline sécrétoire pour la protection des surfaces muqueuses; 4) l’IgE, immunoglobuline fixée sur les mastocytes ou les granulocytes basophiles, impliquée dans les réactions d’hypersensibilité immédiate; 5) l’IgD, dont la fonction essentielle est identique à celle d’un récepteur sur les lymphocytes B.
Immunité à médiation cellulaire. La voie cellulaire du système immunitaire spécifique est sous la dépendance des lymphocytes T. Ces cellules possèdent également des récepteurs antigéniques sur leurs membranes. Elles reconnaissent l’antigène s’il est présenté par les cellules possédant l’antigène (CPA) dans le contexte de l’histocompatibilité antigénique. Ces cellules présentent donc cette limite qui vient s’ajouter à leur spécificité antigénique. Les cellules T, qui agissent comme cellules auxiliaires dans diverses réponses immunitaires (dont la réponse humorale), interviennent dans le recrutement des cellules inflammatoires et peuvent, en tant que cellules T cytotoxiques, tuer les cellules cibles après reconnaissance spécifique de l’antigène.
La résistance de l’hôte dépend de l’intégrité fonctionnelle du système immunitaire, ce qui suppose que les composants cellulaires et moléculaires orchestrant la réponse immunitaire soient présents en quantité suffisante et sous une forme opérationnelle. Les immunodéficiences congénitales chez l’humain se traduisent souvent par des anomalies de certaines souches de lignées cellulaires qui ne produisent pas du tout de cellules immunitaires ou en produisent en nombre insuffisant. Par analogie avec les maladies immunodéficientes humaines congénitales et acquises, l’immunosuppression induite par un produit chimique peut simplement être due au fait que le nombre de cellules fonctionnelles n’est pas aussi élevé qu’il devrait l’être (PISSC, 1996). Ces deux phénomènes, absence de lymphocytes ou présence en nombre réduit, peut avoir des effets plus ou moins graves sur le statut immunitaire. Certains états immunodéficients ou une immunosuppression sévère, tels qu’on peut les observer en transplantation ou après un traitement cytostatique, sont responsables en particulier d’une incidence élevée d’infections opportunistes ou de pathologies néoplasiques. Les infections peuvent être bactériennes, virales, fongiques ou dues à des protozoaires, le type prédominant de l’infection dépendant de l’immunodéficience en cause. L’exposition à des produits chimiques immunosuppresseurs de l’environnement peut être à l’origine de formes plus discrètes d’immunosuppression, plus difficiles à détecter, pouvant conduire, par exemple, à une incidence accrue d’infections telles que la grippe ou le simple rhume.
Etant donné la complexité du système immunitaire et sa très grande diversité de cellules, de médiateurs et de fonctions constituant un réseau élaboré et interactif, les produits immunotoxiques ont de multiples possibilités d’exercer un effet. On ne connaît pas encore parfaitement la nature des lésions initiales induites par de nombreux produits chimiques immunotoxiques, mais on dispose chaque jour d’un peu plus d’informations sur les modifications immunobiologiques à l’origine d’une dépression de la fonction immunitaire, surtout grâce aux études sur les animaux de laboratoire (Dean et coll., 1994). Des effets toxiques peuvent se produire au niveau des fonctions critiques ci-après (nous illustrons le propos grâce à quelques produits immunotoxiques):
On peut dire de l’allergie qu’il s’agit d’un effet indésirable pour la santé résultant de l’induction excessive d’une réponse immunitaire spécifique. Lorsque des réactions d’hypersensibilité se produisent sans la participation du système immunitaire, on parle de pseudo-allergie. Dans le contexte de l’immunotoxicologie, l’allergie est due à une réponse immunitaire spécifique à des produits chimiques ou à des médicaments. Le pouvoir de sensibilisation d’un produit chimique chez l’individu est généralement lié à sa capacité de liaison covalente aux protéines de l’organisme. Les réactions allergiques peuvent être de plusieurs types, qui diffèrent selon le mécanisme immunologique sous-jacent et la vitesse de la réaction. On distingue quatre types principaux de réactions allergiques: les réactions d’hypersensibilité de type I, sous la dépendance d’anticorps IgE, avec une apparition des symptômes quelques minutes après exposition de l’individu sensibilisé; les réactions d’hypersensibilité de type II, dues à l’altération ou à la destruction des cellules hôtes par des anticorps, les symptômes apparaissant dans ce cas en quelques heures; les réactions d’hypersensibilité de type III, ou phénomène d’Arthus, sont également sous la dépendance d’anticorps, mais vis-à-vis d’un antigène soluble et résultent de l’action locale ou systémique de complexes immuns; les réactions de type IV ou d’hypersensibilité retardée sont commandés par les lymphocytes T et, chez un sujet sensibilisé, les symptômes se développent de vingt-quatre à quarante-huit heures après l’exposition.
Les deux types d’allergie chimique les plus importants pour la santé en milieu de travail sont la sensibilisation de contact ou allergie cutanée et l’allergie respiratoire.
Hypersensibilité de contact. Un grand nombre de produits chimiques peuvent être à l’origine d’une sensibilisation de contact. Après une exposition topique d’un sujet sensible à un allergène chimique, il se produit une réponse lymphocytaire T dans les ganglions lymphatiques de drainage. Au niveau de la peau, l’allergène réagit directement ou indirectement avec les cellules épidermiques de Langerhans qui transportent le produit chimique vers les ganglions lymphatiques et le présentent sous une forme immunogène aux lymphocytes T sensibles. Les lymphocytes T activés par l’allergène prolifèrent, provoquant une expansion clonale. L’individu est alors sensibilisé et répondra à une seconde exposition de la peau au même produit chimique par une réaction immunitaire plus agressive, responsable d’un eczéma allergique de contact. La réaction inflammatoire cutanée caractéristique de ce type d’eczéma résulte de la reconnaissance de l’allergène au niveau du tissu cutané par des lymphocytes T spécifiques. Ces lymphocytes deviennent actifs, sécrètent des cytokines et provoquent l’accumulation locale d’autres leucocytes mononucléaires. Les symptômes se développent dans les vingt-quatre à quarante-huit heures suivant l’exposition d’un individu sensible: l’eczéma allergique de contact représente donc une forme d’hypersensibilité de type retardé. Les causes habituelles de l’eczéma allergique de contact sont des produits chimiques organiques (tels que le 2,4-dinitrochlorobenzène), des métaux (tels que le nickel et le chrome) ou des produits d’origine végétale (comme l’urushiol du sumac vénéneux).
Hypersensibilité respiratoire. L’hypersensibilité respiratoire est généralement considérée comme une réaction d’hypersensibilité de type I. Cependant, les réactions de phase tardive et les symptômes chroniques associés à l’asthme paraissent impliquer des processus immunitaires de type cellulaire (type IV). Les symptômes aigus associés à l’allergie respiratoire sont dus aux anticorps IgE, dont la production est provoquée par l’exposition du sujet sensible à l’allergène chimique inducteur. Les anticorps IgE sont distribués par voie systémique et se lient, par l’intermédiaire des récepteurs membranaires, aux mastocytes présents dans les tissus vascularisés, en particulier dans le tractus respiratoire. Lorsque le même produit est inhalé de nouveau, une réaction d’hypersensibilité respiratoire se déclenche. L’allergène se lie à une protéine et se fixe en même temps aux anticorps IgE portés par les mastocytes. Suivent alors une dégranulation des mastocytes et la sécrétion des médiateurs de l’inflammation tels que l’histamine et les leucotriènes. Ces médiateurs sont responsables de la bronchoconstriction et de la vasodilatation, donnant lieu aux symptômes de l’allergie respiratoire: asthme ou rhinite. Parmi les produits chimiques connus pour induire chez l’humain une hypersensibilité respiratoire, il faut citer les anhydrides d’acides (comme l’anhydride trimellitique), certains diisocyanates (TDI, par exemple), les sels de platine et des colorants. L’exposition chronique au béryllium est également connue pour provoquer une pathologie pulmonaire d’hypersensibilité.
On peut dire de l’auto-immunité qu’il s’agit de la stimulation de réponses immunitaires spécifiques dirigées contre les antigènes endogènes du «soi». L’auto-immunité induite peut résulter d’une perturbation de l’équilibre des lymphocytes T régulateurs, ou de la liaison d’un xénobiotique avec des composants tissulaires normaux les rendant immunogènes (altération du «soi»). Les médicaments et produits chimiques connus pour induire fortuitement ou exacerber une maladie auto-immune chez les individus sensibles sont des composés de faible poids moléculaire (100 à 500 daltons) généralement considérés comme non immunogènes. Le mécanisme d’une maladie auto-immune après exposition à un produit chimique est en grande partie inconnu. La maladie peut être provoquée directement par un anticorps circulant, résulter indirectement de la formation de complexes immuns, ou être la conséquence d’une réaction immunologique de type cellulaire. Il s’agit selon toute vraisemblance d’un mécanisme multiple. On connaît mieux le mécanisme pathogène des troubles hémolytiques immunitaires induits par les médicaments:
Des réponses de type auto-immunitaire ont été observées en présence de produits chimiques divers et surtout de médicaments (Kammüller, Bloksma et Seinen, 1989). En milieu de travail, l’exposition à des produits chimiques peut parfois conduire à des syndromes de caractère auto-immunitaire, celle à des produits comme le chlorure de vinyle monomère, le trichloroéthylène, le perchloroéthylène, les résines époxy ou la poussière de silice pouvant provoquer des syndromes du type sclérodermie. L’exposition à l’hydrazine a été associée à un syndrome s’apparentant au lupus érythémateux aigu disséminé. On a aussi constaté que l’exposition au TDI pouvait induire un purpura thrombocytopénique et que les métaux lourds tels que le mercure avaient provoqué des cas de glomérulonéphrite à complexes immuns.
L’évaluation du statut immunitaire chez l’humain s’effectue en étudiant des points de vue quantitatif et fonctionnel, principalement sur le sang périphérique, les immunoglobulines et le complément, ainsi que les sous-populations leucocytaires. Ces méthodes sont généralement identiques à celles que l’on utilise pour étudier l’immunité à médiation humorale et cellulaire, ou la résistance non spécifique, chez des malades présentant une immunodéficience congénitale. Pour les études épidémiologiques (sur des populations exposées professionnellement), les paramètres doivent être sélectionnés sur la base de leur valeur prédictive dans une population humaine, de leur validation par des modèles animaux et des indicateurs biologiques correspondants (voir tableau 33.6). La stratégie de dépistage des effets immunotoxiques après une exposition (accidentelle) à des polluants environnementaux ou à d’autres toxiques dépend davantage des circonstances, comme le type d’immunodéficience attendu, le temps écoulé entre l’exposition et l’évaluation du statut immunitaire, le degré d’exposition et le nombre d’individus exposés. L’évaluation du risque immunotoxique d’un xénobiotique particulier chez l’humain est une démarche extrêmement difficile quand elle n’est pas impossible, en raison surtout de la présence d’un mélange de facteurs d’origine endogène ou exogène qui interviennent dans la réponse des individus à une lésion toxique. C’est vrai en particulier pour les études sur le rôle d’une exposition chimique dans les maladies auto-immunes, où les facteurs génétiques jouent un rôle déterminant.
|
Catégorie de test |
Caractéristiques |
Tests spécifiques |
|
Tests généraux de base à inclure dans un groupe de tests |
Indicateurs de santé et de l�état fonctionnel des organes |
Urée sanguine, glycémie, etc. |
|
Tests immunologiques de base à inclure dans un groupe de tests |
Indicateurs généraux du statut immunitaire |
Numération sanguine complète |
|
Tests spécialisés à inclure en fonction des observations cliniques, des expositions suspectées ou des résultats des tests antérieurs |
Indicateurs de fonctions ou d�événements immunitaires spécifiques |
Génotype d�histocompatibilité |
|
Recherche |
Indicateurs de fonctions ou d�événements immunitaires généraux ou spécifiques |
Tests de stimulation in vitro |
Comme on dispose rarement des données humaines dont on aurait besoin, on évalue la plupart du temps les risques d’immunosuppression induite par un produit chimique chez l’humain en s’appuyant sur des études animales. Dans le cas des xénobiotiques, on détermine leur potentiel immunotoxique surtout grâce à des études contrôlées chez les rongeurs. Les études d’exposition in vivo sont, à cet égard, la meilleure solution en raison de la nature multifactorielle complexe du système immunitaire et des réponses immunes. Les études in vitro sont de plus en plus utilisées pour élucider un mécanisme d’immunotoxicité. De plus, l’étude des effets d’un produit sur des cellules d’origine humaine et animale permet d’acquérir des données utiles pour comparer les espèces. Ces données peuvent alors être utilisées dans une approche de type «parallélogramme» afin d’améliorer l’évaluation du risque. Si on dispose de données pour trois des côtés du parallélogramme (animal in vivo, animal et humain in vitro), il est alors plus facile de prévoir le résultat pour le dernier, c’est-à-dire le risque chez l’humain.
Lorsque l’évaluation d’un risque d’immunosuppression induite par un produit chimique repose uniquement sur des données animales, on a la possibilité pour extrapoler à l’humain d’employer une technique qui consiste à appliquer un facteur d’incertitude à la NOAEL (No Observed Adverse Effect Level (niveau sans effet nocif observé)). On peut se fonder pour cela sur des paramètres établis dans des modèles pertinents, tels que les essais de résistance de l’hôte et l’évaluation in vivo des réactions d’hypersensibilité et de production d’anticorps. Dans l’idéal, il faut valider la pertinence de cette démarche d’évaluation du risque par des études chez l’humain. De telles études devraient associer l’identification et le dosage du toxique, des données épidémiologiques et une évaluation du statut immunitaire.
Pour prévoir une hypersensibilité de contact, on dispose depuis les années soixante-dix de modèles sur cobaye. Bien que sensibles et reproductibles, ces tests ont des limites, car ils reposent sur une évaluation subjective; des méthodes plus récentes et mieux quantifiées développées chez la souris permettent de pallier cette carence. S’agissant de l’hypersensibilité due aux produits chimiques induite par inhalation ou ingestion d’allergènes, il conviendrait de mettre au point des tests et d’évaluer leur valeur prédictive chez l’humain. Lorsqu’on se propose d’établir des niveaux de sécurité pour des allergènes potentiels en milieu de travail, il ne faut pas perdre de vue que l’allergie est un phénomène biphasique: il y a la phase de sensibilisation et la phase de déclenchement. Ainsi, la concentration requise pour provoquer une réaction allergique chez un individu préalablement sensibilisé est beaucoup plus faible que celle qui est nécessaire chez un individu «neuf» du point de vue immunologique, mais sensible.
Comme il n’existe pratiquement pas de modèles animaux pour prévoir l’auto-immunité due aux produits chimiques, il serait bon de s’employer à en mettre au point. Il faut pour cela apprendre à mieux connaître l’auto-immunité induite par les produits chimiques chez l’humain, en particulier grâce à l’étude de marqueurs génétiques et immunologiques permettant d’identifier les individus sensibles. Les sujets exposés aux médicaments induisant une auto-immunité offrent cette possibilité.
L’étude et la caractérisation des propriétés toxiques des produits chimiques et autres agents reposent souvent sur les organes et systèmes atteints. Dans ce chapitre, nous étudierons de manière approfondie le système immunitaire et le gène. Si nous avons porté notre choix sur ces deux cibles, c’est parce qu’elles constituent pour l’une un modèle de cible systémique complexe, pour l’autre un modèle de cible moléculaire. Le lecteur qui souhaite des informations détaillées sur la toxicologie au niveau des organes cibles pourra se reporter à des traités de toxicologie comme ceux de Casarett et Doull, ou encore de Hayes. Le Programme international sur la sécurité des substances chimiques (PISSC) a également publié plusieurs documents de référence classés par système.
Les études de toxicologie au niveau des organes cibles sont entreprises d’ordinaire au vu des informations sur les effets toxiques spécifiques d’une substance, obtenues à partir de données épidémiologiques ou d’études générales de toxicité aiguë ou chronique, ou dans le but de protéger certaines fonctions, par exemple la reproduction ou le développement fœtal. Dans certains cas, les autorités compétentes exigent que des tests spécifiques de toxicité sur des organes cibles soient effectués: c’est le cas aux Etats-Unis où la loi sur les pesticides exige d’effectuer une étude de neurotoxicité (voir l’article «L’approche américaine de l’évaluation du risque des toxiques pour la reproduction et des agents neurotoxiques»), ou au Japon où des dispositions concernant les tests de mutagénicité sont prévues dans la loi sur le contrôle des substances chimiques (voir l’article «Les principes d’identification du risque: l’approche japonaise»).
Comme nous l’expliquons dans l’article «L’organe cible et les effets critiques», l’identification d’un organe critique consiste à déceler l’organe ou le système où un effet nocif s’observe en premier, ou celui qui répond le premier aux doses ou aux expositions les plus faibles. Cette information est ensuite exploitée pour concevoir des investigations toxicologiques spécifiques ou des tests de toxicité mieux orientés capables de mettre en évidence les signes de toxicité les plus sensibles au niveau de l’organe cible. Les études de toxicologie au niveau de ces organes peuvent également servir à préciser les mécanismes d’action, nécessaires à l’évaluation du risque (voir l’article «L’approche américaine de l’évaluation du risque des toxiques pour la reproduction et des agents neurotoxiques»).
L’étude des organes cibles peut se faire en exposant des organismes intacts et en analysant très précisément la fonction et l’histopathologie de l’organe cible, ou en maintenant en culture in vitro pendant des périodes plus ou moins longues des cellules, des coupes tissulaires ou d’organes entiers (voir l’article «Les mécanismes de la toxicologie: introduction et concepts»). Il arrive dans certains cas que l’on dispose de tissus d’origine humaine pour ce type d’études de toxicité. Il est alors possible de valider les hypothèses d’extrapolation interespèces. Cependant, on ne doit pas oublier que de telles études ne fournissent pas d’informations sur les toxicocinétiques respectives.
En général, les études de toxicité au niveau des organes cibles comportent toutes les étapes suivantes: examen histopathologique détaillé de l’organe cible, y compris post mortem, poids tissulaire, examen des tissus après fixation; études biochimiques de voies critiques dans l’organe cible, telles que les systèmes enzymatiques importants; études fonctionnelles de l’aptitude de l’organe et des constituants cellulaires à assumer des fonctions spécifiques métaboliques ou autres; étude des indicateurs biologiques d’exposition et des effets précoces dans les cellules de l’organe cible.
L’étude des organes cibles peut aussi avoir pour finalité de chercher à mieux connaître leur physiologie, leur biochimie et leur biologie moléculaire. Or, comme la synthèse et la sécrétion de protéines de faible poids moléculaire sont des aspects importants de la fonction rénale, les études de néphrotoxicité s’y intéressent souvent (PISSC, 1991). De même, étant donné que les communications intercellulaires constituent un processus fondamental du fonctionnement du système nerveux, les études des organes cibles en neurotoxicité comportent des mesures neurochimiques et biophysiques détaillées de la synthèse, de la captation, du stockage, de la libération et de la liaison au récepteur des neurotransmetteurs ainsi que des mesures électrophysiologiques des modifications des potentiels de membrane qui leur sont associées.
Comme on s’efforce par tous les moyens de ne plus employer les animaux de laboratoire ou de moins les utiliser, on s’emploie à mettre au point des méthodes d’études in vitro de toxicité au niveau des organes cibles. Des progrès considérables ont été accomplis en ce sens avec les toxiques des fonctions de reproduction (Heindell et Chapin, 1993).
En résumé, les études de toxicité sur les organes cibles sont généralement envisagées comme des études spécialisées de toxicologie. Le choix des organes cibles pour ce type d’évaluation est orienté par les résultats des tests de dépistage, tels que les tests de toxicité aiguë ou subaiguë utilisés dans les pays de l’OCDE et de l’Union européenne; certains organes cibles et systèmes sont a priori candidats pour ce type d’investigation en raison de leur intérêt dans la prévention d’effets nocifs pour la santé.
Un indicateur biologique, ou marqueur biologique, est un paramètre mesurable présent dans un système biologique, par exemple l’organisme humain. Ce paramètre est considéré comme étant le reflet, ou le marqueur, de l’état général de l’organisme ou de son espérance de vie. Dans le domaine de la santé au travail, les indicateurs biologiques sont d’ordinaire utilisés comme indicateurs de l’état de santé ou du risque de survenue d’une pathologie.
Les indicateurs biologiques servent aussi bien dans les études in vitro que dans les études in vivo, l’humain pouvant être inclus dans celles-ci. On distingue habituellement trois types d’indicateurs biologiques: les indicateurs d’exposition, d’effet ou de sensibilité/réceptivité (voir tableau 33.7). Il est à noter que certains indicateurs sont difficilement classables.
|
Echantillon |
Mesure |
Objectif |
|
Indicateurs biologiques d�exposition |
||
|
Tissu adipeux |
Dioxine |
Exposition à la dioxine |
|
Sang |
Plomb |
Exposition au plomb |
|
Os |
Aluminium |
Exposition à l�aluminium |
|
Air expiré |
Toluène |
Exposition au toluène |
|
Cheveux |
Mercure |
Exposition au méthylmercure |
|
Sérum |
Benzène |
Exposition au benzène |
|
Urine |
Phénol |
Exposition au benzène |
|
Indicateurs biologiques d�effet |
||
|
Sang |
Carboxyhémoglobine |
Exposition au monoxyde de carbone |
|
Hématies |
Protoporphyrine-zinc |
Exposition au plomb |
|
Sérum |
Cholinestérase |
Exposition aux organo-phosphorés |
|
Urine |
Microglobulines |
Exposition à un néphrotoxique |
|
Leucocytes |
Adduits à l�ADN |
Exposition à un mutagène |
Sous réserve d’un degré de validité acceptable, les indicateurs biologiques peuvent avoir plusieurs finalités. A l’échelle individuelle, un indicateur biologique peut servir à valider ou à récuser un diagnostic d’intoxication ou d’effets indésirables liés à un produit chimique. Chez un sujet en bonne santé, il peut traduire une hypersensibilité individuelle à l’exposition à un produit chimique donné et permettre ainsi de prévoir le risque et d’envisager des mesures de prévention. Dans un groupe de travailleurs exposés, certains indicateurs biologiques d’exposition peuvent être utilisés pour évaluer si la réglementation antipollution est effectivement respectée ou encore dans quelle mesure les efforts de prévention générale sont efficaces.
Un indicateur biologique d’exposition peut impliquer un xénobiotique (ou son métabolite) présent dans l’organisme, un produit d’interaction entre le xénobiotique (ou son métabolite) et un constituant endogène, ou un autre paramètre lié à l’exposition. Plus fréquemment, les indicateurs biologiques d’exposition pour des produits stables tels que les métaux consistent à mesurer la concentration de ces produits dans des échantillons appropriés: sang, sérum ou urine. Dans le cas des produits chimiques volatils inhalés avec l’air contaminé, on peut évaluer leur concentration dans l’air exhalé. Si le produit est métabolisé dans l’organisme, on peut choisir un ou plusieurs métabolites comme indicateurs biologiques d’exposition, les métabolites étant souvent dosés dans des échantillons urinaires.
Les techniques modernes d’analyse permettent de séparer les isomères ou les homologues de produits organiques, de déterminer la spéciation des produits métalliques ou les isotopes de certains éléments. Des méthodes très perfectionnées permettent d’établir les modifications que la fixation de produits chimiques réactifs fait subir à la structure de l’ADN ou d’autres macromolécules. Ces nouvelles techniques d’étude des indicateurs biologiques sont sans nul doute appelées à se développer, car elles permettent d’améliorer les limites de détection et la validité analytique.
Des progrès très prometteurs ont été réalisés dans le cas des produits chimiques mutagènes. Ces produits, qui sont des intermédiaires réactifs, peuvent former des adduits avec les macromolécules, telles que protéines ou ADN. Les adduits à l’ADN sont mis en évidence dans les leucocytes ou les biopsies tissulaires. Des fragments spécifiques d’ADN peuvent être excrétés dans l’urine. Ainsi, une exposition à l’oxyde d’éthylène est responsable de l’élimination urinaire de N-(2-hydroxyéthyl)guanine, du fait de la réaction de l’oxyde d’éthylène avec l’ADN, après excision des bases endommagées. Certains adduits peuvent ne pas être directement reliés à une exposition précise. Par exemple, la 8-hydrox-2´-désoxyguanosine, témoin d’un processus oxydatif sur l’ADN, doit sa formation à plusieurs produits chimiques, la plupart d’entre eux agissant également comme inducteurs de peroxydation lipidique.
D’autres macromolécules sont également susceptibles d’être modifiées par formation d’adduits ou par oxydation. En particulier, les produits intermédiaires réactifs peuvent entraîner la formation d’adduits à l’hémoglobine que l’on peut utiliser comme indicateurs biologiques d’exposition. L’avantage est qu’on peut obtenir une quantité importante d’hémoglobine à partir d’un échantillon sanguin et que, comme les hématies ont une durée de vie de quatre mois, les adduits formés avec les acides aminés de l’hémoglobine permettent d’évaluer l’exposition totale durant cette période.
Les adduits peuvent être mesurés par des techniques sensibles telles que la chromatographie liquide à haute performance ou des méthodes immunologiques. En général, ces méthodes analytiques récentes sont coûteuses et nécessitent d’être améliorées et validées. Une meilleure sensibilité est obtenue en utilisant la technique du postmarquage au 32P, qui est une mesure non spécifique des lésions de l’ADN. Toutes ces techniques trouvent leur application en surveillance biologique et ont du reste été employées dans de nombreuses études. Cependant, on a besoin de méthodes analytiques plus simples et plus sensibles. Etant donné la spécificité limitée de certaines de ces méthodes pour les expositions de faible niveau, certains facteurs tels que le tabagisme peuvent avoir une incidence significative sur les résultats des mesures et poser de ce fait des difficultés d’interprétation.
L’exposition à des produits mutagènes ou à des composés métabolisés en produits mutagènes peut être évaluée grâce à l’analyse des urines. Un échantillon prélevé chez un sujet exposé est mis à incuber avec une souche bactérienne chez laquelle une mutation ponctuelle spécifique s’exprime d’une manière facile à mesurer. Si des produits chimiques mutagènes sont présents dans l’échantillon urinaire, un taux accru de mutations s’observe chez la bactérie.
Les indicateurs biologiques d’exposition doivent être évalués en tenant compte de la variation de la durée de l’exposition et des relations entre les divers compartiments. Ainsi, l’échelle de temps représentée par l’indicateur biologique, autrement dit la mesure dans laquelle l’indicateur biologique peut refléter les expositions passées ou la charge corporelle accumulée, doit être déterminée à partir de données toxicocinétiques afin d’interpréter le résultat. En particulier, on doit prendre en compte la façon dont l’indicateur biologique reflète l’intensité de la rétention d’un produit au niveau des organes cibles spécifiques. Les échantillons sanguins sont souvent utilisés dans les études d’indicateur biologique; cependant, le sang périphérique n’est généralement pas considéré comme un compartiment en soi, bien qu’il serve de transporteur entre les compartiments. La relation entre la concentration sanguine et le niveau atteint dans les différents organes varie de manière importante en fonction du produit chimique, de la durée de l’exposition et du temps écoulé depuis la fin de l’exposition.
Ce type de raisonnement est parfois utilisé pour classer un indicateur biologique comme indicateur de dose (totale) absorbée ou comme indicateur de dose efficace (c’est-à-dire de la quantité qui a atteint le tissu cible). Par exemple, l’exposition à un solvant donné peut être évaluée par le dosage de sa concentration sanguine à un moment donné après l’exposition. Ce dosage reflète alors la quantité de solvant absorbée par l’organisme. En raison de sa pression de vapeur, une partie est exhalée. Pendant qu’il circule par voie sanguine, le solvant réagit avec divers composants de l’organisme et subit éventuellement une dégradation enzymatique. Ces processus métaboliques peuvent être estimés en dosant les acides mercapturiques produits par conjugaison avec le glutathion. L’excrétion cumulée de ces acides reflétera mieux la dose efficace que la concentration sanguine.
Des événements de la vie, tels que la reproduction ou la sénescence, sont susceptibles de modifier la distribution d’un produit chimique. Ainsi, au cours de la grossesse, de nombreux produits chimiques peuvent franchir la barrière placentaire, provoquant ainsi l’exposition du fœtus. La lactation entraîne l’excrétion de produits chimiques liposolubles, ce qui diminue la rétention chez la mère et augmente la captation chez l’enfant. En cas de perte de poids ou d’ostéoporose, les produits chimiques stockés peuvent être libérés et causer une augmentation et une prolongation de l’exposition «endogène» des organes cibles. D’autres facteurs, pour lesquels on dispose d’indicateurs biologiques de sensibilité, peuvent modifier l’absorption, le métabolisme, la rétention et la distribution d’un produit chimique chez un individu (voir ci-après).
Un indicateur biologique d’effet peut être un composant endogène, une mesure de capacité fonctionnelle ou tout autre indice de l’état ou de l’équilibre de l’organisme ou d’un organe, modifié par une exposition. Les indicateurs biologiques d’effet sont généralement des indices d’anomalies précliniques.
Ils peuvent être spécifiques ou non. Les indicateurs biologiques spécifiques peuvent être utilisés à des fins préventives, car ils permettent de relier un effet biologique donné à une exposition définie. Les indicateurs biologiques non spécifiques ne permettent pas de relier un effet à une cause particulière, car ils peuvent refléter l’effet global intégré en relation avec des expositions composites. Ces deux types d’indicateurs biologiques peuvent ainsi être très utiles en santé au travail.
Il n’existe pas de distinction claire entre les indicateurs biologiques d’exposition et les indicateurs biologiques d’effet. On peut dire par exemple que la formation d’adduits est plus un indicateur d’effet qu’un indicateur d’exposition. Cependant, les indicateurs biologiques d’effet correspondent en général à des altérations de fonctions au niveau de cellules, de tissus ou même de l’organisme dans son ensemble. Certains chercheurs considèrent comme indicateurs biologiques d’effet des modifications importantes, comme une hépatomégalie chez les animaux de laboratoire exposés ou une diminution de croissance chez les enfants. En ce qui concerne la santé au travail, les indicateurs biologiques d’effet devraient être limités à ceux qui permettent de révéler des modifications biochimiques subcliniques ou réversibles (une inhibition enzymatique, par exemple). L’indicateur biologique d’effet le plus fréquemment utilisé est certainement l’inhibition des cholinestérases provoquée par certains insecticides, nommément les organophosphorés et les carbamates. Dans la plupart des cas, cet effet est totalement réversible et l’inhibition de l’enzyme reflète l’exposition globale à ce groupe particulier d’insecticides.
Certaines expositions n’entraînent pas une inhibition enzymatique, mais plutôt une induction. C’est le cas de plusieurs enzymes appartenant à la famille du cytochrome P450 (voir l’article «Les déterminants génétiques de la réponse toxique») qui peuvent être induites à la suite de l’exposition à certains solvants et hydrocarbures aromatiques polycycliques. Comme ces enzymes s’expriment surtout dans des tissus dont il est difficile d’obtenir une biopsie, l’activité enzymatique est déterminée in vivo de façon indirecte en administrant au sujet un produit qu’elles métabolisent puis en dosant le produit dégradé dans l’urine ou le plasma.
D’autres expositions peuvent induire la synthèse d’une protéine protectrice. Le meilleur exemple de ce phénomène est sans doute celui de la métallothionéine, qui fixe le cadmium et assure l’excrétion de ce métal; l’exposition au cadmium est l’un des facteurs qui provoquent une augmentation de l’expression du gène de la métallothionéine. Il existe d’autres protéines protectrices du même type, mais elles n’ont pas encore été suffisamment étudiées pour être considérées comme des indicateurs biologiques. Parmi les candidats possibles, on trouve les protéines dites de stress, appelées anciennement protéines du choc thermique. Ces protéines sont produites par toute une gamme d’organismes en réponse à diverses expositions nocives.
Les lésions oxydatives peuvent être évaluées par le dosage de la concentration sérique du malondialdéhyde ou celle de l’éthane dans l’air expiré. De même, l’excrétion urinaire de protéines de faible poids moléculaire, l’albumine par exemple, peut être utilisée comme indicateur biologique précoce d’une lésion rénale. Plusieurs paramètres utilisés en routine clinique (comme les taux d’hormones ou les activités enzymatiques sériques) peuvent aussi constituer des indicateurs biologiques précieux. Cependant, beaucoup de ces paramètres ne sont pas suffisamment sensibles pour permettre la détection d’une lésion précoce.
Un autre groupe d’indicateurs d’effet concerne les effets génotoxiques (modifications de structure des chromosomes). Ces effets peuvent être mis en évidence par examen au microscope des leucocytes en période de division. Des lésions importantes sur les chromosomes — aberrations chromosomiques ou formation de micronoyaux — sont en effet observables au microscope. Ces lésions peuvent également être observées à l’aide d’un colorant ajouté aux cellules pendant leur division. On peut ainsi visualiser l’exposition à un agent génotoxique par une augmentation de l’échange de colorant entre les deux chromatides de chaque chromosome (test des échanges de chromatides sœurs (ECS)). Si les aberrations chromosomiques sont le signe d’un risque accru de développer un cancer, le test des ECS est de signification moins nette.
La génotoxicité peut aussi être évaluée grâce à une technique plus complexe reposant sur l’existence de mutations ponctuelles dans des cellules somatiques, notamment les leucocytes ou les cellules épithéliales de la muqueuse buccale. Une mutation sur un locus spécifique peut rendre les cellules capables de croître dans un milieu de culture contenant un produit chimique normalement toxique (par exemple, la 6-thioguanine). On peut également évaluer le produit spécifique d’un gène (comme les concentrations sériques ou tissulaires d’oncoprotéines codées par des oncogènes particuliers). Bien entendu, ces mutations reflètent une lésion génotoxique globale et ne permettent pas d’attribuer la cause à une exposition particulière. Ces méthodes ne sont pas encore suffisamment au point pour avoir une utilisation pratique en santé au travail, mais de rapides progrès dans cette voie de recherche laissent penser qu’elles seront disponibles dans quelques années.
Un indicateur de sensibilité, héréditaire ou induite, est un indice permettant de déterminer si un sujet est particulièrement sensible à l’effet d’un xénobiotique ou d’un groupe de xénobiotiques. La sensibilité d’origine génétique a suscité beaucoup d’intérêt, alors que d’autres facteurs sont au moins aussi importants. L’hypersensibilité peut être la conséquence d’un facteur héréditaire, être liée à l’environnement ou encore être constitutionnelle.
L’aptitude à métaboliser certains produits chimiques varie d’un individu à l’autre en fonction de facteurs génétiques (voir l’article «Les déterminants génétiques de la réponse toxique»). Plusieurs enzymes importantes paraissent être contrôlées par un seul gène. Par exemple, l’oxydation des xénobiotiques est assurée principalement par une famille d’enzymes appartenant au cytochrome P450. D’autres enzymes se chargent de rendre les métabolites plus hydrosolubles par conjugaison (par exemple, la N-acétyltransférase et la µ-glutathion-S-transférase). L’activité de ces enzymes, qui est contrôlée génétiquement, varie considérablement. Comme mentionné précédemment, l’activité peut être évaluée en administrant une petite dose d’un médicament puis en mesurant la quantité de métabolite dans l’urine. Certains des gènes correspondants sont maintenant bien identifiés, et on dispose de techniques pour déterminer le génotype. D’importantes études suggèrent que le risque de développer certaines formes de cancer est en relation avec la capacité de métaboliser des xénobiotiques. De nombreuses questions restent encore sans réponse, ce qui limite à l’heure actuelle l’utilisation de ces indicateurs biologiques de sensibilité en santé au travail.
D’autres facteurs héréditaires, comme les déficiences en alpha1-antitrypsine ou en glucose-6-phosphate déshydrogénase, sont également responsables d’une diminution des mécanismes de défense, expliquant une hypersensibilité à certaines expositions.
Pour l’essentiel, la recherche relative à la sensibilité s’est intéressée à la prédisposition génétique alors que d’autres facteurs qui jouent pourtant un rôle ont été négligés. Ainsi, les individus ayant une maladie chronique peuvent être plus sensibles à une exposition professionnelle. De même, lorsqu’une maladie en cours ou une exposition antérieure à un produit toxique ont provoqué des lésions organiques subcliniques, la résistance à une nouvelle exposition toxique est assurément diminuée. Des paramètres biochimiques peuvent dans ce cas être utilisés comme indicateurs biologiques de sensibilité. Les réponses allergiques constituent sans doute le meilleur exemple d’hypersensibilité. Lorsqu’un individu est sensibilisé à une exposition particulière, on peut déceler chez lui des anticorps spécifiques au niveau sérique. Même si le sujet n’est pas devenu sensible, les expositions actuelles ou antérieures font augmenter le risque de développer un effet indésirable s’il est soumis à une exposition professionnelle.
L’évaluation de l’effet additif des expositions à des mélanges en milieu professionnel représente un défi de taille. De plus, les habitudes personnelles et la prise de médicaments peuvent accroître la sensibilité. Par exemple, la fumée de tabac contient généralement une quantité importante de cadmium. Ainsi, en cas d’exposition professionnelle au cadmium, un gros fumeur qui a accumulé dans son organisme des quantités importantes de ce métal présente un risque accru de développer une maladie rénale liée au cadmium.
Les indicateurs biologiques sont extrêmement utiles en recherche toxicologique et beaucoup peuvent être utilisés en surveillance biologique. Il faut cependant en connaître les limites. De nombreux indicateurs biologiques ont été étudiés uniquement chez les animaux de laboratoire. Un modèle toxicocinétique valable pour une espèce ne l’est pas nécessairement chez l’humain, et avant de procéder à des extrapolations à ce dernier, il est indispensable de confirmer les résultats par des études sur des volontaires. On doit prendre aussi en compte les variations individuelles dues à des facteurs génétiques ou constitutionnels.
Dans certains cas, les indicateurs biologiques d’exposition sont impossibles à utiliser (comme dans le cas des produits chimiques ayant une durée de vie in vivo très courte). Certains de ces produits peuvent s’accumuler et provoquer des lésions dans des organes inaccessibles aux techniques de routine, par exemple le système nerveux. La distribution d’un produit dans l’organisme, et de ce fait la mesure et l’interprétation d’un indicateur biologique, peuvent aussi varier en fonction de la voie d’exposition. Ainsi, une exposition cérébrale par voie olfactive directe échappe à toute possibilité de détection par indicateurs biologiques d’exposition. S’agissant des indicateurs biologiques d’effet, beaucoup d’entre eux ne sont pas spécifiques, pour des raisons très diverses, dont le mode de vie. L’interprétation doit être extrêmement prudente à l’heure actuelle en ce qui concerne les indicateurs biologiques de sensibilité, car on ne connaît pas encore l’incidence des génotypes individuels sur la santé.
En santé au travail, l’indicateur biologique idéal devrait satisfaire à plusieurs exigences. Le recueil des échantillons et l’analyse doivent tout d’abord être simples et fiables. Les indicateurs doivent être normalisés pour assurer une qualité analytique optimale en tenant compte des conditions spécifiques qui sont très variables. Parmi les difficultés à surmonter, il faut citer la préparation du sujet, la procédure d’échantillonnage, la manipulation des échantillons et la technique de dosage; celle-ci comporte des facteurs techniques, comme le calibrage ou les procédures d’assurance qualité, et des variables individuelles telles que le niveau d’instruction et la formation des techniciens de laboratoire.
En ce qui concerne la validité analytique et la traçabilité des résultats, les matériaux de référence doivent être choisis de façon pertinente, avec des concentrations appropriées en substances toxiques ou en métabolites significatifs. Les laboratoires chargés de l’évaluation des indicateurs utilisés en surveillance biologique ou à des fins diagnostiques doivent employer des procédures analytiques bien documentées, aux performances bien définies, et archiver les résultats pour qu’il soit facile de les vérifier. L’aspect financier des matériaux de référence utilisés pour assurer la qualité ne doit pas non plus être oublié. La qualité des résultats obtenus et l’usage qui en est fait doivent être mis en équilibre avec le surcoût de l’assurance qualité, qui inclut les matériaux de référence, la main-d’œuvre et le matériel.
Un indicateur biologique doit satisfaire à une autre exigence: il doit être spécifique, du moins dans les conditions de l’étude et pour un type particulier d’exposition. L’indicateur biologique choisi doit permettre d’établir une relation claire avec le degré d’exposition pour éviter tout problème d’interprétation. De plus, si on veut pouvoir interpréter correctement un résultat, il est essentiel de bien connaître son intérêt diagnostique (autrement dit, la traduction de la valeur de l’indicateur biologique en intensité de risque éventuel pour la santé). Dans ce domaine, les métaux servent de paradigme pour les indicateurs biologiques. Les recherches récentes ont montré combien la relation dose-réponse est complexe et subtile et combien il est difficile de déterminer des niveaux sans effet et, par conséquent, de fixer une exposition admissible. Ces recherches ont néanmoins eu le mérite d’indiquer le type d’investigation et les améliorations nécessaires pour parvenir à des informations utiles. Les relations quantitatives entre exposition et effets nocifs pour la santé ne sont pas encore connues pour la plupart des produits organiques; dans de nombreux cas, les organes cibles primaires ne sont pas établis de façon certaine. De plus, l’évaluation des données sur la toxicité et les concentrations des indicateurs biologiques est souvent compliquée parce que le sujet n’est pas exposé à un seul produit, mais à un mélange de substances.
Avant qu’on puisse utiliser un indicateur biologique en santé au travail, il faut répondre à un certain nombre d’exigences. Tout d’abord, il faut que l’indicateur reflète uniquement une modification subclinique et encore réversible. Ensuite, étant donné que les résultats d’un indicateur biologique peuvent être interprétés en terme de risque pour la santé, on doit disposer de moyens de prévention et être en mesure de les appliquer si les résultats indiquent qu’il est nécessaire de réduire l’exposition. Enfin, l’utilisation pratique d’un indicateur biologique doit être considérée comme acceptable du point de vue de l’éthique.
Les résultats des mesures effectuées en hygiène du travail peuvent être comparées aux limites d’exposition applicables. De même, les résultats des indicateurs biologiques d’exposition ou d’effet peuvent être comparés aux limites d’activité biologique, parfois assimilées à des indices biologiques d’exposition. Ces limites doivent être fondées sur les avis éclairés de cliniciens et de scientifiques des disciplines concernées, et les administrateurs responsables, en leur qualité de «gestionnaires du risque», devraient prendre en compte les facteurs éthiques, sociaux, culturels et économiques pertinents. La base scientifique doit, si possible, inclure les relations dose-réponse complétées par des informations sur les variations de sensibilité dans la population soumise au risque. Dans certains pays, les travailleurs et des représentants de la société civile participent de façon active à l’établissement des normes, surtout lorsque les données scientifiques sont insuffisantes. L’une des principales incertitudes réside dans la définition des effets nocifs à éviter. Peut-on dire, par exemple, que la formation d’adduits en tant qu’indicateur biologique d’exposition représente un tel effet nocif (c’est-à-dire un indicateur biologique d’effet) qui doit être prévenu? Il est difficile du point de vue de l’éthique de décider s’il est défendable de fixer, pour un même produit, des limites différentes dans le cas d’une exposition fortuite ou d’une exposition professionnelle.
L’information concernant l’utilisation des indicateurs biologiques doit être transmise dans le cadre de la relation médecin-patient aux individus suivis. Il faut tenir compte en particulier des questions éthiques que posent les indicateurs biologiques expérimentaux qui ne sont pas interprétables actuellement du point de vue des risques réels pour la santé. Ainsi, en dehors des valeurs normales de la plombémie, les directives qui existent en ce qui concerne l’interprétation des indicateurs biologiques d’exposition à l’échelle de la population générale sont limitées. La confiance dans les données obtenues est également importante. Il faut se demander si l’échantillonnage a été bien fait et si le laboratoire a appliqué des procédures dûment validées d’assurance qualité. Autre domaine de préoccupation: celui de l’hypersensibilité individuelle. Toutes ces données doivent être prises en compte avant d’exploiter les résultats d’une étude.
Les secteurs de la société touchés ou intéressés par la réalisation d’une étude d’indicateur biologique doivent tous pouvoir participer aux décisions devant être prises sur la façon d’utiliser l’information qui en ressort. Des procédures spécifiques pour prévenir ou surmonter les conflits éthiques inévitables doivent être prévues dans les structures légales et sociales de la région ou du pays. Cependant, chaque situation est particulière du fait des questions qu’elle soulève et des pièges qu’elle comporte, et le problème de la participation de la société civile ne saurait être résolu par une procédure unique qui engloberait toutes les applications des indicateurs biologiques d’exposition.
L’évaluation de la toxicité génétique est l’étude de la faculté qu’ont certains agents d’induire, au niveau du matériel génétique (ADN), des lésions ou des mutations de l’un des trois types suivants: génique, chromosomique ou génomique. Chez l’être humain, les gènes sont composés d’ADN, ensemble constitué d’unités appelées bases nucléotidiques, et sont organisés en structures physiques appelées chromosomes. La génotoxicité peut entraîner des effets significatifs et irréversibles sur la santé humaine. Les lésions génotoxiques constituent une étape critique dans l’induction d’un cancer et sont également responsables de malformations congénitales et de mort fœtale. Les trois types de mutations mentionnées ci-dessus peuvent se produire au niveau des cellules germinales ou somatiques.
Les tests utilisés dans le domaine des mutations géniques permettent de déceler trois types d’effet: la substitution, l’addition ou la délétion de nucléotides à l’intérieur d’un gène. Ceux utilisés pour détecter des mutations chromosomiques mettent en évidence les cassures ou les réarrangements chromosomiques impliquant un ou plusieurs chromosomes. Les tests portant sur les mutations génomiques décèlent les modifications du nombre de chromosomes ou aneuploïdie. L’évaluation de la toxicité génétique a considérablement évolué avec la mise au point en 1927, par Hermann Müller, du premier test de détection d’agents génotoxiques (mutagènes). Depuis, plus de 200 tests ont été mis au point pour déceler les mutations sur l’ADN, mais on en utilise aujourd’hui moins d’une dizaine de façon courante. Le présent article dresse un inventaire de ces tests, du type de mesures qu’ils permettent d’effectuer et explique leur rôle dans l’évaluation de la toxicité.
La toxicologie génétique fait maintenant partie intégrante du processus d’évaluation du risque en général et son pouvoir prédictif en termes de risque cancérogène a récemment gagné en importance. Cependant, avant l’avènement de la toxicologie génétique (avant 1970), on employait d’autres méthodes, et on le fait encore, pour identifier le risque cancérogène potentiel chez l’humain. Il faut citer ici les études épidémiologiques, les études in vivo à long terme, les études in vivo à moyen terme, les études in vivo et in vitro à court terme, l’étude des relations structure-activité (intelligence artificielle) et les études basées sur le mécanisme d’action.
Le tableau 33.8 présente les avantages et les inconvénients de ces diverses méthodes.
|
|
Avantages |
Inconvénients |
|
Etudes épidémiologiques |
1) l�humain est l�indicateur ultime de la maladie; |
1) caractère généralement rétrospectif (actes de décès, etc.); |
|
Etudes in vivo à long terme |
1) évaluation prospective et rétrospective (validation); |
1) rarement reproduites, moyens nécessaires importants; |
|
Etudes in vivo et in vitro à court et moyen terme |
1) plus rapides et moins coûteuses que les autres études; |
1) données in vitro n�assurant pas une bonne prédiction des données in vivo; |
|
Associations structure |
1) assez faciles, rapides, et peu coûteuses; |
1) non �biologiques�; |
|
Déductions basées sur le mécanisme d�action |
1) assez exactes pour certaines catégories de produits chimiques; |
1) mécanismes de cancérogenèse chimique non définis, multiples et probablement spécifiques d�un produit chimique ou d�une catégorie; |
Bien que le type et le nombre de techniques employées pour évaluer la toxicité génétique soient en évolution constante et varient d’un pays à l’autre, les plus utilisées font appel à des tests: 1) de mutation génique chez les bactéries ou sur des cultures de cellules de mammifères; 2) de mutation chromosomique sur des cultures de cellules de mammifères ou sur moelle osseuse de souris in vivo. Certains des tests de la seconde catégorie peuvent également détecter une aneuploïdie. Même s’ils ne décèlent pas les mutations germinales, ces tests n’en sont pas moins utilisés surtout en raison du coût élevé et de la complexité des méthodes sur cellules germinales. Malgré ces deux inconvénients, on recourt cependant aux tests sur cellules germinales de souris lorsqu’on désire obtenir une information sur les effets au niveau de ces cellules.
Des études systématiques sur vingt-cinq ans (1970-1995), effectuées en particulier dans le cadre du Programme national américain de toxicologie en Caroline du Nord, ont permis de conclure à l’utilité d’un petit nombre de tests pour mettre en évidence une activité mutagène. L’intérêt d’un test était établi sur la base de son aptitude à détecter des agents cancérogènes chez les rongeurs et donc susceptibles de provoquer un cancer chez l’humain. Ainsi, les études de ces dernières décennies ont montré que les cellules cancéreuses portent des mutations sur certains gènes et que de nombreux cancérogènes sont également mutagènes. On considère les cellules cancéreuses comme des cellules somatiques portant des mutations, et la cancérogenèse comme un type de mutagenèse de cellules somatiques.
Les tests de toxicité génétique utilisés le plus couramment aujourd’hui ont été sélectionnés non seulement en raison de l’importance de leur base de données, de leur coût relativement faible et de leur facilité d’exécution, mais aussi parce qu’ils ont permis de détecter de nombreux cancérogènes chez les rongeurs et, on le présume, chez l’humain. Les tests de toxicité génétique servent donc à prévoir le pouvoir cancérogène potentiel des agents chimiques.
La toxicologie génétique a fait un progrès important sur les plans conceptuel et pratique lorsqu’on s’est aperçu que les enzymes de l’organisme modifient de nombreux cancérogènes en les transformant en formes dégradées (métabolites) qui, bien souvent, représentent la forme mutagène et cancérogène ultime du produit chimique initial. Heinrich Malling a montré que l’inclusion d’une préparation de foie de rongeur, qui apporte la plupart des enzymes nécessaires à cette transformation ou activation métabolique, permet de reproduire ce métabolisme en boîte de Pétri. De nombreux tests de toxicologie génétique réalisés en boîtes ou en tubes (in vitro) utilisent donc un ajout de préparations enzymatiques similaires. Les préparations simples sont appelées mélanges S9, et les préparations purifiées, microsomes. Des bactéries ou des cellules de mammifères génétiquement modifiées contiennent certains gènes humains ou des gènes de rongeurs les rendant capables de produire ces enzymes, ce qui supprime la nécessité d’employer le mélange S9 ou des microsomes.
Les systèmes bactériens primaires qui servent au dépistage de la toxicité génétique sont les tests de mutagenèse sur Salmonella (Ames) et, dans une moindre mesure, sur la souche WP2 d’Escherichia coli. Les études réalisées aux alentours de 1985 montrent qu’il suffit d’utiliser deux souches du type Salmonella (TA98 et TA100) pour déceler 90% environ des mutagènes connus chez Salmonella. Aussi ces deux souches sont-elles employées pour la plupart des dépistages, même si on peut faire appel à d’autres souches pour une détection plus approfondie.
Ces tests sont réalisés de diverses façons, mais les deux procédés de base sont les tests d’inclusion en boîte de Pétri et de suspension en phase liquide. Dans le test d’inclusion en boîte de Pétri, les cellules, le produit chimique testé et (si nécessaire) le mélange S9 sont ajoutés ensemble à de l’agarose liquéfié et coulés à la surface d’une boîte de Pétri. La couche d’agarose supérieure durcit en quelques minutes et les boîtes sont mises à incuber pendant deux à trois jours; après ce temps, les cellules mutantes ont poussé pour former des groupes de cellules visibles à l’œil nu ou colonies, qui sont alors comptées. L’agarose contient des agents sélectifs ou des composants tels que seules les cellules nouvellement mutées seront susceptibles d’y croître. Le test par incubation en phase liquide est similaire, mais les cellules, le produit testé et le mélange S9 sont mis à incuber ensemble dans une phase liquide ne contenant pas d’agarose liquéfié; les cellules sont ensuite lavées pour éliminer le produit testé et le mélange S9, puis ensemencées sur de l’agarose.
Les mutations sur cultures de cellules de mammifères sont étudiées essentiellement sur l’un des deux gènes: hprt et tk. De même que pour les tests bactériens, les lignées cellulaires de mammifères (développées à partir de cellules de rongeurs ou de cellules humaines) sont exposées au produit à étudier dans des boîtes de culture en plastique ou dans des tubes, puis sont ensemencées dans des boîtes de culture contenant un milieu avec un agent sélectif qui permet aux seules cellules mutantes de se développer. Les tests employés sont les tests CHO/HPRT, TK6 et L5178Y/TK+/– du lymphome de souris. On utilise aussi d’autres lignées cellulaires présentant diverses mutations au niveau de la réparation de l’ADN ou contenant certains gènes humains participant au métabolisme. Ces systèmes permettent une réversion des mutations à l’intérieur du gène (mutation génique) ainsi que des mutations impliquant les régions du chromosome encadrant le gène (mutation chromosomique). Néanmoins, ce dernier type de mutation concerne davantage le système tk que le système hprt du fait de la localisation du gène tk.
Comme pour les tests d’incubation en phase liquide pour la mutagenèse bactérienne, les tests de mutagenèse sur cellules de mammifères exigent généralement qu’on expose des cellules (dans des boîtes de culture ou dans des tubes) au produit à étudier en présence du mélange S9 pendant plusieurs heures. Les cellules sont ensuite lavées et cultivées pendant quelques jours supplémentaires afin de permettre aux produits du gène normal (type sauvage) d’être dégradés et aux produits du gène nouvellement muté d’être exprimés et accumulés, puis elles sont ensemencées dans un milieu contenant un agent sélectif qui permet aux seules cellules mutantes de croître. Comme pour les tests bactériens, les cellules mutantes se développent en colonies visibles à l’œil nu qui sont ensuite comptées.
La mutation chromosomique est mise en évidence principalement par des tests cytogénétiques qui consistent à exposer des rongeurs, des cellules de rongeurs ou des cellules humaines en boîtes de culture à un produit chimique pour le tester. On laisse s’écouler le temps nécessaire à une ou à plusieurs divisions cellulaires et on colore les chromosomes avant de les examiner au microscope pour détecter les modifications de structure ou du nombre des chromosomes. Bien que de nombreuses anomalies puissent être observées, les plus couramment retenues par les agences réglementaires comme étant les plus fiables sont les aberrations chromosomiques et le test du micronoyau.
L’examen des cellules présentant des aberrations chromosomiques exige un bon entraînement et une grande compétence, ce qui rend cette procédure coûteuse et longue. Au contraire, le test du micronoyau requiert peu d’expérience, et la détection peut être automatisée. Les micronoyaux apparaissent comme de petites taches ponctuelles à l’intérieur de la cellule, distinctes du noyau où se trouvent les chromosomes. Ils résultent soit d’une cassure de chromosome, soit d’une aneuploïdie. Comme ils sont plus faciles à observer que les aberrations chromosomiques, et comme des études récentes ont montré que les agents causant des aberrations chromosomiques dans la moelle osseuse de souris in vivo induisent en général des micronoyaux dans ce tissu, il est maintenant courant de les mesurer pour déterminer l’aptitude d’un produit à provoquer une mutation chromosomique.
Bien que les tests sur cellules germinales soient utilisés beaucoup moins fréquemment que les tests décrits ci-dessus, ils sont indispensables pour établir si un produit représente un risque pour les cellules germinales, des mutations au niveau de ces cellules pouvant entraîner des effets sur la santé des générations suivantes. Les tests sur cellules germinales les plus communément utilisés le sont chez la souris et supposent l’emploi de systèmes permettant la détection de: 1) translocations (échanges) héréditaires entre chromosomes (test de translocation héréditaire); 2) mutations géniques ou chromosomiques impliquant des gènes spécifiques (tests du locus spécifique, visuels ou biochimiques); 3) mutations affectant la viabilité (test du dominant létal). Comme pour les tests sur cellules somatiques, l’utilisation des tests sur cellules germinales repose sur l’hypothèse de travail que les produits donnant une réponse positive dans ces tests sont des mutagènes potentiels pour les cellules germinales humaines.
Des études récentes effectuées sur 41 produits cancérogènes pour les rongeurs (cancérogènes et mutagènes somatiques potentiels pour l’humain) ont montré qu’il suffit de disposer de trois types d’information pour en caractériser environ 90%: 1) la connaissance de la structure chimique du produit, en particulier le fait qu’il soit électrophile (voir l’article «La relation structure-activité»); 2) les tests de mutagenèse sur Salmonella; 3) un test de toxicité chronique sur 90 jours chez le rongeur (souris et rat). De fait, le caractère mutagène de la totalité des cancérogènes humains recensés par le CIRC peut être mis en évidence en utilisant uniquement le test sur Salmonella et le test du micronoyau sur moelle osseuse de souris. L’utilisation de ces tests de mutagenèse pour la détection des cancérogènes humains potentiels se justifie d’autant plus que la plupart des cancérogènes humains sont cancérogènes chez la souris et le rat (cancérogènes transespèces) et que la plupart des cancérogènes transespèces sont mutagènes sur Salmonella ou induisent des micronoyaux au niveau de la moelle osseuse de souris.
Avec les progrès réalisés en technologie de l’ADN, dans la connaissance du génome humain et dans la compréhension du rôle des mutations dans le cancer, il est probable que des tests de génotoxicité en cours de développement seront intégrés aux protocoles standards de détection. Il faut citer en particulier les tests sur cellules ou rongeurs transgéniques. Les systèmes transgéniques sont des systèmes où un gène d’une espèce différente est introduit dans une cellule ou un organisme. Par exemple, des souris transgéniques, obtenues après introduction d’un gène bactérien, sont maintenant utilisées de façon expérimentale pour déceler une mutation dans un organe ou un tissu de l’animal. On dispose actuellement de cellules bactériennes telles que Salmonella et de cellules de mammifères (y compris des lignées cellulaires humaines) contenant des gènes participant au métabolisme d’agents cancérogènes/mutagènes, tels que les gènes du cytochrome P450. Il est ainsi possible d’effectuer l’analyse moléculaire des mutations induites au niveau du transgène chez le rongeur transgénique, ou dans les gènes natifs tels que hprt, ou dans les gènes cibles chez Salmonella, ce qui permet de déterminer la nature exacte des mutations induites par les produits chimiques et de donner un aperçu du mécanisme d’action du produit chimique tout en facilitant les comparaisons avec les mutations chez l’humain susceptible d’être exposé à l’agent.
Les avancées moléculaires en cytogénétique permettent désormais une évaluation plus fine des mutations chromosomiques. On peut citer l’utilisation de sondes (petits morceaux d’ADN) qui s’attachent (s’hybrident) à des gènes spécifiques. Les réarrangements des gènes sur le chromosome peuvent alors être révélés par la modification de localisation des sondes, facilement visualisées en raison de leur fluorescence. L’électrophorèse sur gel pour mettre en évidence les cassures d’ADN (communément appelée test des «comètes») permet leur détection dans une cellule isolée et peut être un outil extrêmement précieux si on l’associe aux techniques cytogénétiques pour déceler des lésions chromosomiques.
Après de nombreuses années d’utilisation et de développement systématique d’une importante base de données, on peut maintenant procéder à l’évaluation de la toxicité génétique avec quelques tests seulement, pour un coût relativement faible et en peu de temps (quelques semaines). Les données obtenues peuvent être utilisées pour prévoir le caractère cancérogène d’un produit chez les rongeurs ou potentiellement cancérogène ou mutagène somatique chez l’humain. Grâce à cette possibilité, on est en mesure de limiter l’introduction dans l’environnement d’agents mutagènes et cancérogènes et de développer des produits de remplacement non mutagènes. Les études futures devraient conduire à des méthodes encore plus perfectionnées ayant un pouvoir prédictif supérieur aux méthodes actuellement utilisées.
L’émergence de techniques sophistiquées en biologie moléculaire et cellulaire a favorisé une rapide évolution des sciences de la vie et en particulier de la toxicologie. Dans la pratique, cette évolution s’est traduite par un déplacement du centre d’intérêt de la toxicologie qui se consacre non plus à l’animal entier ou à des groupes d’animaux, mais aux cellules ou aux molécules issues d’un seul sujet, qu’il soit animal ou humain. Depuis le milieu des années quatre-vingt, les toxicologues emploient ces nouvelles méthodes pour évaluer les effets de produits chimiques sur les systèmes vivants. Selon une progression logique, ces techniques se sont adaptées aux objectifs de la toxicologie expérimentale et ont évolué à la fois en fonction de ces progrès scientifiques et des préoccupations d’ordre économique et sociologique.
Compte tenu du nombre important de substances à tester, l’aspect économique est déterminant. Une pléthore de nouveaux produits cosmétiques, pharmaceutiques, pesticides, chimiques et ménagers, dont il faut évaluer la toxicité potentielle, sont mis chaque année sur le marché. Ajoutons à cela que de très nombreux produits d’utilisation courante n’ont pas encore été correctement testés. Le recueil d’informations détaillées sur la sécurité de tous ces produits chimiques grâce aux méthodes traditionnelles d’étude sur animal entier serait extrêmement coûteux et long, en admettant même que cette tâche titanesque soit réalisable.
Notre société est de plus en plus exigeante face aux problèmes de santé publique et de sécurité et la population remet en question le bien-fondé de l’emploi des animaux pour tester les produits chimiques. S’agissant de la sécurité des produits chimiques pour l’humain, le public et les écologistes exercent une pression soutenue sur les instances gouvernementales pour qu’elles appliquent une réglementation plus rigoureuse. On peut citer ici le mouvement qu’ont lancé les écologistes aux Etats-Unis pour obtenir l’interdiction du chlore et des composés chlorés en arguant que la plupart de ces composés n’avaient jamais été testés de manière suffisante. Du point de vue toxicologique, le fait d’interdire une classe entière de produits chimiques uniquement parce qu’ils contiennent du chlore est à la fois irresponsable et non rationnel scientifiquement. Cependant, on peut comprendre que le public veuille s’assurer que les produits chimiques libérés dans l’environnement ne présentent pas de risque pour la santé. Cet exemple illustre bien la nécessité d’employer des méthodes plus efficaces et plus rapides pour évaluer la toxicité.
Autre problème de société dont les conséquences sont importantes pour les études toxicologiques: le bien-être des animaux. De par le monde, les associations de protection des animaux sont de plus en plus nombreuses à s’opposer farouchement à l’utilisation d’animaux vivants pour les études de sécurité chimique. Elles ont organisé des campagnes actives contre les fabricants de produits cosmétiques, ménagers ou pharmaceutiques pour faire interdire les expérimentations animales. En Europe, ces efforts ont abouti à l’adoption du sixième amendement de la directive 76/768/CEE (directive sur les produits cosmétiques). Aux termes de cette directive, les produits et les ingrédients cosmétiques testés sur des animaux vivants ne pourront plus être vendus dans l’Union européenne après le 1er janvier 1998, sauf lorsqu’il n’existe pas de solution de remplacement suffisamment validée. Cette directive ne s’applique pas aux produits commercialisés sur le marché américain et dans d’autres pays, mais elle aura néanmoins des répercussions considérables sur les sociétés internationales qui commercialisent ce type de produits, notamment en Europe.
Le concept de solutions de remplacement, base du développement des tests autres que ceux réalisés sur animal entier, est défini par la règle dite des trois R: réduction du nombre d’animaux utilisés; raffinement des protocoles, afin que les animaux soient soumis à moins de stress et d’inconfort; et remplacement des tests actuels sur animal entier par des tests in vitro (c’est-à-dire des tests qui ne sont pas réalisés sur des animaux vivants), ou par des modèles informatiques, ou encore par des tests sur vertébrés inférieurs ou invertébrés. Cette théorie des trois R a été présentée pour la première fois en 1959 dans un ouvrage publié par deux scientifiques britanniques, W.M.S. Russell et Rex Burch, The Principles of Humane Experimental Technique. Pour ces deux chercheurs, seul un traitement humain des animaux permet d’obtenir des résultats scientifiques valables. Ils proposaient pour cela de développer des méthodes propres à réduire le nombre d’animaux nécessaires à ces expériences dans le but ultime de les remplacer. Il est intéressant de noter que les principes énoncés par Russell et Burch ont eu peu d’écho au début et qu’il a fallu attendre le milieu des années soixante-dix pour qu’on assiste à la renaissance du mouvement en faveur de la protection des animaux de laboratoire. Aujourd’hui, le principe des trois R est bien ancré dans les domaines de la recherche, de l’expérimentation et de l’éducation.
On peut dire pour résumer que le développement des méthodologies in vitro résulte d’une série de facteurs qui ont convergé vers le même but au cours des deux dernières décennies. Il est difficile de dire si un seul de ces facteurs aurait permis d’exercer une telle pression sur la stratégie de l’évaluation toxicologique.
Dans cette section, il est uniquement question des méthodes in vitro d’évaluation de la toxicité qui sont destinées à remplacer l’expérimentation sur animal entier. Les autres méthodes de substitution telles que les modèles informatiques ou les relations quantitatives structure-activité sont abordées dans d’autres parties du présent chapitre.
Les études in vitro sont généralement conduites sur des cellules ou des tissus d’origine animale ou humaine. L’expression in vitro, qui signifie littéralement «dans du verre», renvoie aux expériences réalisées sur du matériel vivant ou des composants de matériel vivant cultivés en boîtes de Pétri ou dans des tubes à essai dans des conditions bien définies. Ces procédures s’opposent aux études in vivo réalisées «sur des animaux vivants». Bien qu’il soit difficile, sinon impossible, de prévoir les effets d’un produit chimique sur un organisme complexe lorsqu’on limite l’observation à un seul type de cellules dans une boîte, les études in vitro livrent une grande quantité d’informations sur la toxicité intrinsèque d’un produit ou sur son mécanisme de toxicité cellulaire et moléculaire. De plus, elles offrent de nombreux avantages sur les études in vivo, car elles sont généralement moins coûteuses et peuvent être réalisées dans des conditions mieux contrôlées. En outre, bien qu’on ait encore besoin d’un petit nombre d’animaux vivants pour obtenir les cellules nécessaires et les cultiver in vitro, on peut néanmoins considérer que ces méthodes constituent des solutions de remplacement puisqu’elles répondent aux exigences de réduction (utilisation d’un petit nombre d’animaux par rapport aux études in vivo) et de raffinement (les animaux ne sont plus soumis aux effets toxiques observés lors des expériences in vivo).
Pour interpréter les résultats d’un test de toxicité in vitro, décider s’il présente de l’intérêt aux fins de l’évaluation de la toxicité et le rapporter au processus toxicologique général in vivo, il est nécessaire de savoir quelle partie du processus toxicologique ce test permet d’étudier. Ce processus comporte en effet une succession d’événements qui débutent par l’exposition de l’organisme à un agent physique ou chimique et donnent lieu à des interactions cellulaires et moléculaires qui finalement se manifestent dans la réponse de l’organisme entier. Les tests in vitro sont généralement limités à la partie du processus toxicologique qui se produit aux niveaux cellulaire et moléculaire. Les informations recueillies à partir de telles études vont de la voie métabolique et de l’interaction de métabolites actifs avec des cibles cellulaires et moléculaires aux paramètres toxiques potentiellement quantifiables qui peuvent servir d’indicateurs biologiques moléculaires de l’exposition. Dans l’idéal, il faut connaître le mécanisme toxique d’un produit chimique depuis l’exposition jusqu’aux manifestations de l’organisme, si l’on veut que les informations obtenues lors de tests in vitro puissent être interprétées et mises en relation avec la réponse de l’organisme entier. Cependant, c’est pratiquement impossible, puisque les mécanismes toxicologiques qui ont été totalement élucidés sont assez rares. Les toxicologues se trouvent donc dans la situation où les résultats d’un test in vitro ne permettent pas de prévoir exactement la toxicité in vivo puisque les mécanismes toxicologiques restent inconnus. Il existe cependant quelques cas où un test in vitro permet d’élucider le(s) mécanisme(s) cellulaires et moléculaires de toxicité.
Reste un problème majeur concernant le développement et la réalisation de tests in vitro: doivent-ils être fondés sur le mécanisme d’action ou suffit-il qu’ils soient descriptifs? Du point de vue scientifique, il est de loin préférable d’employer des tests in vitro fondés sur le mécanisme d’action. L’espoir de développer, dans un avenir proche, un test in vitro capable de remplacer totalement un test sur animal entier est pratiquement nul, si on ne connaît pas parfaitement le mécanisme d’action. Mais cela ne doit pas exclure l’utilisation de tests descriptifs comme outils de dépistage précoce, ce qui est le cas actuellement. Ces tests in vitro ont abouti à une réduction significative du nombre d’animaux utilisés et, tant que le mécanisme d’action n’est pas bien connu, il peut s’avérer nécessaire d’employer, de façon plus limitée, des tests in vitro dont les résultats sont en bonne corrélation avec ceux que l’on peut obtenir in vivo.
Nous décrirons dans cette section plusieurs tests in vitro élaborés pour évaluer le potentiel cytotoxique d’un produit chimique. Ils sont, pour la plupart, faciles à réaliser et automatisables. L’un d’eux, qu’on emploie couramment, est le test au rouge neutre. Ce test est réalisé sur des cultures de cellules: dans la plupart des applications, les cellules sont placées dans des boîtes de culture comportant 96 puits de 6,4 mm de diamètre. Chaque puits ne pouvant être utilisé que pour une seule détermination, cette disposition permet de tester en double échantillon de multiples concentrations du produit chimique avec des contrôles positifs et négatifs. Après traitement des cellules par des concentrations du produit chimique à tester croissantes d’au moins deux ordres de grandeur (de 0,01 mM à 1 mM, par exemple) et par des produits chimiques contrôles positifs et négatifs, les cellules sont rincées et traitées au rouge neutre, colorant absorbé et retenu par les cellules vivantes uniquement. Le colorant peut être ajouté immédiatement après élimination du produit chimique testé pour déterminer les effets immédiats, ou à des intervalles variables après son élimination pour préciser les effets cumulatifs ou différés. L’intensité de la coloration correspond au nombre de cellules vivantes dans chaque puits. On utilise pour cela un spectrophotomètre équipé d’un lecteur de plaque programmé qui mesure l’intensité de chacun des 96 puits de la boîte de culture. Cette méthode automatisée permet de réaliser rapidement une étude concentration-réponse et d’obtenir des données statistiques.
Une autre méthode d’étude de la cytotoxicité relativement simple est le test au MTT. Le MTT (3[4,5-diméthylthiazol-2-yl]-2,5-diphényltétrazolium bromure) est un colorant de type tétrazolium qui est réduit par les enzymes mitochondriales en un composé coloré en bleu. Seules les cellules dont les mitochondries sont viables vont donner cette réaction; en conséquence, l’intensité de la couleur est directement proportionnelle au degré d’intégrité des mitochondries. Il s’agit d’un test utile pour détecter des composés cytotoxiques en général ainsi que les agents ayant les mitochondries pour cible spécifique.
La mesure de l’activité de la lactico-déshydrogénase (LDH) est également très utilisée pour étudier la cytotoxicité. Cette enzyme, normalement présente dans le cytoplasme des cellules vivantes, est libérée dans le milieu de culture lorsque les membranes cellulaires sont lésées par un agent toxique. Des prélèvements de petites quantités de milieu de culture effectués à différents moments après le traitement chimique des cellules permettent de mesurer la quantité de LDH libérée dans le milieu et de suivre le déroulement de la toxicité en fonction du temps. Bien que la libération de LDH constitue une évaluation très générale de la cytotoxicité, elle n’en est pas moins utile parce qu’elle est facile à réaliser, et ce en temps réel.
De nombreuses méthodes sont mises au point actuellement pour détecter une lésion cellulaire. Les plus complexes font appel à des sondes fluorescentes pour mesurer divers paramètres intracellulaires: libération de calcium, modifications du pH ou du potentiel de membrane. Ces sondes, en général très sensibles, permettent de déceler des modifications cellulaires minimes, bien antérieures à la mort cellulaire. De plus, ces méthodes fluorescentes sont pour la plupart automatisables grâce à l’utilisation des plaques à 96 puits et de lecteurs de plaque.
Après avoir recueilli, par l’une de ces méthodes, des données sur une série de produits chimiques, il est possible de déterminer leur toxicité relative. La toxicité relative d’un agent chimique, dans un test in vitro, est exprimée par la concentration permettant d’obtenir 50% de réponses par rapport à des cellules non traitées. Cette détermination, qui représente la CE50 (concentration efficace pour 50% des cellules), est utilisée pour comparer la toxicité in vitro de différents produits chimiques (on emploie également un terme semblable pour évaluer la toxicité relative, la CI50, qui correspond à la concentration de produit entraînant une inhibition de 50% d’un processus cellulaire, par exemple la capacité d’absorption du rouge neutre). Il n’est pas facile de comparer la toxicité relative in vitro d’un produit chimique à sa toxicité relative in vivo, car de nombreux facteurs de confusion interviennent in vivo, tels que la toxicocinétique, le métabolisme, les mécanismes de réparation ou de défense. De plus, la plupart de ces méthodes mesurent une cytotoxicité générale et ne sont pas basées sur le mécanisme d’action. Par conséquent, il est seulement possible d’établir une corrélation entre les toxicités relatives in vitro et in vivo. En dépit de la complexité et des nombreuses difficultés que présente l’extrapolation des données obtenues in vitro aux fins d’une exploitation in vivo, ces tests in vitro se sont révélés très utiles en raison de leur simplicité et de leur faible coût. Ils peuvent être employés comme tests de dépistage pour détecter les médicaments ou les produits chimiques très toxiques dès le début des travaux de développement.
On peut également utiliser les tests in vitro pour évaluer la toxicité au niveau d’un organe cible. La conception de ces tests pose de nombreuses difficultés, la plus notable étant l’impossibilité de maintenir les particularités de l’organe in vivo au moyen d’un système in vitro. Il arrive très souvent qu’une fois prélevées sur l’animal et mises en cultures, les cellules se dégradent rapidement ou qu’elles se différencient, c’est-à-dire qu’elles perdent les fonctions spécifiques de l’organe pour reprendre un état indifférencié. En très peu de temps, généralement quelques jours, les cultures ne permettent plus d’évaluer les effets toxiques spécifiques d’une substance sur un organe donné.
La plupart de ces problèmes sont en voie d’être réglés grâce aux récentes avancées de la biologie cellulaire et moléculaire. Les informations recueillies sur l’environnement cellulaire in vivo peuvent servir à modifier les conditions de culture in vitro. Depuis le milieu des années quatre-vingt, de nouveaux facteurs de croissance et cytokines ont été découverts qui sont pour la plupart disponibles dans le commerce. Grâce à l’addition de ces facteurs, on parvient maintenant à préserver l’intégrité cellulaire des cultures et à conserver leurs fonctions différenciées pendant des périodes plus longues. Les progrès dans la connaissance des besoins nutritifs et hormonaux des cultures de cellules ont également contribué à mettre au point de nouveaux milieux de culture. Il est maintenant possible de cultiver les cellules sur des matrices extracellulaires naturelles ou artificielles. Ces matrices ont une influence considérable sur la structure et la fonction des cellules en culture. L’un de leurs grands avantages est qu’elles permettent de contrôler de façon précise l’environnement de ces cellules et d’examiner les effets de chacun des facteurs sur les processus cellulaires de base et sur leurs réponses aux différents agents chimiques. En un mot, ces systèmes sont le gage d’une meilleure compréhension des mécanismes de toxicité spécifiques de l’organe.
De nombreuses études de toxicité au niveau d’un organe cible sont conduites sur des cellules primaires, qui sont par définition des cellules fraîchement prélevées sur un organe et ont généralement un temps de vie limité en culture. L’emploi de cultures primaires d’un seul type cellulaire d’un organe pour évaluer une toxicité présente bien des avantages. Du point de vue du mécanisme d’action, ces cultures sont utiles pour étudier la cible cellulaire spécifique d’un produit chimique. Dans certains cas, deux ou plusieurs types cellulaires d’un organe peuvent être cultivés ensemble, ce qui permet en plus l’étude des interactions cellulaires dans la réponse à un toxique. Certains systèmes de coculture de cellules cutanées ont été mis au point pour reproduire une structure tridimensionnelle ressemblant à la peau in vivo. Il est également possible de réaliser une coculture de cellules venant de différents organes, par exemple, foie et rein. Ce type de coculture paraît utile pour évaluer les effets spécifiques sur les cellules rénales d’un produit chimique métabolisé au niveau hépatique.
Les outils de biologie moléculaire ont également joué un rôle important dans le développement de lignées cellulaires continues utiles à l’étude de la toxicité au niveau d’un organe cible. Ces lignées cellulaires sont fabriquées en transfectant l’ADN de cellules primaires. Lors d’une procédure de transfection, les cellules et l’ADN sont traitées de façon que l’ADN soit capté par les cellules. L’ADN est généralement issu d’un virus et contient un ou plusieurs gènes qui, une fois exprimés, rendent les cellules immortelles (c’est-à-dire capables de vivre et de croître en culture pendant de longues périodes). L’ADN peut également être manipulé de sorte que le gène immortalisant soit contrôlé par un promoteur inductible. Grâce à ce type de construction, les cellules se divisent uniquement lorsqu’elles reçoivent le stimulus chimique adéquat pour permettre l’expression du gène immortalisant. Citons à titre d’exemple le grand gène de l’antigène T issu du virus simien no 40 (SV40) (gène immortalisant), précédé par la région promoteur du gène de la métallothionéine, induit par la présence d’un métal dans le milieu de culture. Après transfection de ce gène dans les cellules, leur traitement par de faibles concentrations de zinc stimule le promoteur de la métallothionéine et permet au gène de l’antigène T de s’exprimer, ce qui entraîne une prolifération cellulaire. Lorsque le zinc est éliminé du milieu, les cellules cessent de se diviser et, dans les meilleures conditions, retournent à un état où elles expriment leurs fonctions tissulaires spécifiques.
La possibilité de fabriquer des cellules immortalisées, alliée aux progrès des techniques de culture cellulaire, a fortement contribué à créer des lignées cellulaires de nombreux organes différents, dont le cerveau, le rein et le foie. Cependant, avant de pouvoir utiliser en toute sécurité ces lignées cellulaires comme substituts des lignées cellulaires véritables, on doit étudier avec soin leurs caractéristiques pour s’assurer qu’elles sont bien «normales».
Les autres systèmes in vitro utilisés pour étudier la toxicité au niveau d’un organe cible sont beaucoup plus complexes. Au fur et à mesure que ces systèmes gagnent en complexité, allant de la culture cellulaire simple à celle d’un organe entier, ils se rapprochent des systèmes in vivo, tout en restant beaucoup plus difficiles à contrôler en raison du nombre de variables à maîtriser. Par conséquent, l’avantage que l’on tire de l’augmentation du niveau d’organisation peut être compromis du fait que le chercheur manque de contrôle sur l’environnement expérimental. Le tableau 33.9 compare quelques-unes des caractéristiques des systèmes in vitro utilisés pour l’étude de l’hépatotoxicité.
|
Système |
Complexité (niveau de l�interaction) |
Conservation des fonctions hépatiques spécifiques |
Durée potentielle de la culture |
Contrôle de l�environnement |
|
Lignées cellulaires immortalisées |
En partie intercellulaire |
Faible à bonne |
Illimitée |
Excellent |
|
Cultures primaires d�hépatocytes |
Intercellulaire |
Moyenne à excellente |
De quelques jours à quelques semaines |
Excellent |
|
Cocultures de cellules hépatiques |
Intercellulaire (entre types cellulaires identiques ou différents) |
Bonne à excellente |
Quelques semaines |
Excellent |
|
Coupes de foie |
Intercellulaire (entre les divers types cellulaires) |
Bonne à excellente |
De quelques heures à quelques jours |
Bon |
|
Foie isolé perfusé |
Intercellulaire (entre les divers types cellulaires) et à l�intérieur de l�organe |
Excellente |
Quelques heures |
Moyen |
En expérimentation toxicologique, l’emploi des coupes tissulaires fines est de plus en plus répandu. Le chercheur dispose pour les réaliser de nouveaux instruments permettant d’obtenir des coupes homogènes en milieu stérile. Ces coupes présentent des avantages sur les cultures cellulaires, car les différents types cellulaires de l’organe y sont présents et l’architecture in vivo ainsi que les communications intercellulaires y sont maintenues. Des études in vitro peuvent alors être conduites pour déterminer aussi bien le type cellulaire cible dans un organe que pour rechercher la toxicité spécifique au niveau de l’organe cible. L’inconvénient de ces coupes est qu’elles se dégradent rapidement après vingt-quatre heures de culture, surtout à cause d’une oxygénation insuffisante des cellules dans la partie intérieure des coupes. Cependant, de récentes études ont montré qu’on peut assurer une aération plus efficace par rotation douce. Cette solution, de même que l’utilisation d’un milieu de culture plus complexe, permet aux coupes de survivre jusqu’à quatre-vingt-seize heures.
Pour étudier la toxicité de produits chimiques au niveau des organes cibles spécifiques, on peut aussi se servir d’explants tissulaires, dont le principe s’apparente à celui des coupes tissulaires. Ces explants sont réalisés en prélevant une petite quantité de tissu (un embryon entier dans le cas des études de tératogenèse) et en la mettant en culture pour l’étudier. Les cultures d’explants ont été notamment utilisées pour les études de toxicité à court terme, du type études d’irritation et de corrosion cutanée, études de toxicité de l’amiante sur la trachée ou encore études de neurotoxicité sur le tissu cérébral.
Autre solution: les organes isolés perfusés. Ces organes offrent un avantage comparable à celui des coupes tissulaires et des explants puisque tous les types cellulaires d’un organe y sont présents, sans le stress tissulaire qu’imposent les manipulations pour préparer les coupes. De plus, ils permettent le maintien des interactions à l’intérieur de l’organe. Mais ils présentent un grave inconvénient: ils n’ont qu’une faible viabilité, ce qui limite leur utilisation pour une étude toxicologique in vitro. Du point de vue solution de remplacement, ces cultures peuvent être considérées comme un raffinement puisque les animaux ne souffrent pas des conséquences néfastes du traitement par un toxique in vivo. Leur utilisation ne fait cependant pas diminuer de façon très sensible le nombre de sujets nécessaires.
En résumé, on dispose de plusieurs sortes de systèmes in vitro que l’on peut conjuguer pour évaluer la toxicité au niveau des organes cibles. Une difficulté subsiste: l’extrapolation des résultats obtenus dans un système in vitro, partie relativement mineure d’un processus toxicologique, au processus entier qui a lieu in vivo.
Le test de toxicité sur animal entier sans doute le plus contesté du point de vue du bien-être de l’animal est le test de Draize qu’on utilise pour étudier l’irritation oculaire sur le lapin. Pour cela, on place une dose donnée du produit chimique dans l’un des yeux du lapin, l’autre œil servant de témoin. Le degré d’irritation et d’inflammation est évalué à des intervalles de temps donnés après le début de l’exposition. Des efforts importants sont déployés pour trouver des solutions permettant de remplacer ce test, critiqué non seulement pour des raisons humanitaires, mais aussi à cause de la subjectivité des observations et de la variabilité des résultats. Précisons qu’en dépit des violentes critiques qu’il a suscitées, le test de Draize est d’une remarquable efficacité pour identifier les irritants oculaires pour l’humain, notamment les substances de légèrement à modérément irritantes dont le pouvoir est difficile à établir par d’autres méthodes. Il s’impose donc de développer le plus vite possible des solutions de remplacement in vitro.
La recherche de méthodes susceptibles de remplacer le test de Draize est difficile, même si tout porte à croire qu’elle sera fructueuse. De nombreuses techniques in vitro et d’autres solutions de remplacement ont été développées et, dans certains cas, mises en pratique. Parmi les solutions de raffinement du test de Draize, donc moins douloureuses ou pénibles pour les animaux, on peut citer le test oculaire à faible volume, qui consiste à placer de plus petites quantités du produit testé dans les yeux du lapin, non seulement pour des raisons humanitaires, mais aussi pour reproduire de façon plus réaliste les quantités réelles auxquelles l’individu peut être exposé accidentellement. Autre amélioration: celle qui consiste à ne plus tester sur l’animal des substances dont le pH est inférieur à 2 ou supérieur à 11,5, car on sait qu’elles sont extrêmement irritantes pour l’œil.
Entre 1980 et 1989, on estime que le nombre de lapins utilisés pour tester l’irritation oculaire de produits cosmétiques a diminué de 87%. Cette réduction draconienne des tests sur animal entier s’explique par l’incorporation de tests in vitro dans une approche par étapes. Cette démarche fait appel à un processus à plusieurs étapes qui débute par l’examen minutieux des données historiques du pouvoir irritant oculaire et l’analyse physico-chimique du produit à évaluer. Si ces deux étapes ne fournissent pas assez d’informations, on procède alors à une série de tests in vitro. Les données supplémentaires obtenues à partir des tests in vitro peuvent alors être suffisantes pour évaluer la sécurité de la substance, sinon on réalise en dernier recours des tests in vivo en les limitant le plus possible. On voit qu’on peut ainsi se passer complètement des animaux ou du moins réduire considérablement leur nombre.
La batterie de tests in vitro utilisée dans cette stratégie par étapes dépend des exigences de la branche concernée. Le test d’irritation oculaire est en effet pratiqué dans des branches très diverses, depuis celle des cosmétiques jusqu’aux produits industriels en passant par l’industrie pharmaceutique. Vu la diversité des informations recherchées, il n’est pas possible de définir une batterie unique de tests in vitro. En principe, une batterie de tests sert à évaluer cinq paramètres: la cytotoxicité, les modifications physiologiques et biochimiques tissulaires, la relation quantitative structure-activité, les médiateurs de l’inflammation, la guérison et la réparation. Un exemple de test de cytotoxicité, cause possible d’irritation, est le test au rouge neutre sur cultures de cellules (voir ci-dessus). Les modifications physiologiques et biochimiques cellulaires résultant d’une exposition à un produit chimique peuvent être étudiées sur des cultures de cellules épithéliales de cornée humaine. Comme alternative, les chercheurs ont également utilisé des globes oculaires intacts ou disséqués de bœuf ou de poulet obtenus auprès des abattoirs. Les paramètres mesurés dans ces cultures d’organe entier sont souvent les mêmes que ceux mesurés in vivo, par exemple l’opacité ou le gonflement de la cornée.
L’inflammation oculaire est l’un des troubles fréquents de l’exposition aux produits chimiques. On dispose de nombreux moyens pour l’étudier. Diverses méthodes biochimiques permettent de déceler la présence de médiateurs libérés durant le processus inflammatoire, tels que l’acide arachidonique et les cytokines. La membrane chorio-allantoïdienne d’œuf de poulet peut également être utilisée comme indicateur de l’inflammation. Dans ce test, une petite partie de la coquille d’un embryon de poulet de 10 à 14 jours est enlevée, afin d’appliquer le produit chimique sur la membrane, après quoi on étudie à intervalles les signes d’inflammation, comme l’hémorragie vasculaire.
La guérison et la réparation d’une lésion oculaire font partie des paramètres in vivo les plus difficiles à évaluer in vitro. On dispose d’un nouvel instrument, le microphysiomètre au silicium, qui permet de mesurer de faibles variations du pH extracellulaire et peut être utilisé pour contrôler in situ les cultures cellulaires. Cette étude montre une assez bonne corrélation avec la guérison in vivo et est donc utilisée comme test in vitro. Tel se présente aujourd’hui l’inventaire succinct des tests actuellement employés en lieu et place du test de Draize pour l’irritation oculaire. Il est probable qu’au cours des prochaines années, on parviendra à mettre au point et à valider toute une série de tests in vitro pour chaque type d’utilisation spécifique.
Pour qu’un test in vitro soit agréé par les instances réglementaires, il faut d’abord et avant tout qu’il soit validé. Ce processus permet en effet d’établir sa fiabilité dans un but spécifique. Des efforts ont été entrepris, aux Etats-Unis comme en Europe, pour définir et coordonner les processus de validation. En 1993, l’Union européenne a créé le Centre européen pour la validation des méthodes alternatives afin de coordonner les efforts en Europe et d’agir en association avec des organismes américains tels que le Centre universitaire Johns Hopkins de recherche sur les méthodes alternatives de test sur les animaux (CAAT) et le Comité de coordination interagences pour la validation des méthodes alternatives (ICCVAM), composé de représentants des Instituts nationaux de la santé (NHI), de l’Agence américaine de protection de l’environnement (EPA), de l’Administration américaine de réglementation des denrées alimentaires et des produits pharmaceutiques (Food and Drug Administration (FDA)) et de la Commission pour la sécurité des produits de consommation (Consumer Products Safety Commission).
La validation des tests in vitro requiert une organisation et une planification considérables pour que les instances réglementaires et les scientifiques s’entendent sur des procédures acceptables. Il faut aussi qu’un organisme consultatif scientifique assure un contrôle suffisant et garantisse que les protocoles satisfont à certaines normes. Les études de validation devraient être réalisées dans des laboratoires de référence utilisant des agents chimiques homologués distribués par une banque unique de produits chimiques, de cellules et de tissus. Lorsqu’on cherche à faire agréer un test, il faut apporter la preuve, au moyen d’une analyse statistique sérieuse, que ses résultats sont reproductibles au sein d’un même laboratoire et d’un laboratoire à l’autre. Une fois que les résultats des différents volets des études de validation ont été rassemblés, l’organisme consultatif peut se prononcer sur la validité du(es) test(s). De plus, les résultats des études devraient être diffusés dans des publications révisées par des pairs et être consignés dans des bases de données.
La définition du processus de validation est engagée dans la bonne voie. Chaque nouvelle étude de validation fournit des informations utiles à l’étude suivante. La communication et la coopération internationales sont essentielles au développement rapide d’une série de protocoles auxquels de nombreuses personnes pourront souscrire, étant donné en particulier l’urgence imposée par l’adoption de la directive sur les cosmétiques de la Communauté européenne. Cette mesure législative peut en effet donner l’élan nécessaire à un effort de validation sérieux. C’est seulement à l’issue de ce processus que les instances réglementaires pourront envisager d’accepter les méthodes in vitro.
Cet article dresse un bilan succinct de l’état actuel de l’expérimentation toxicologique in vitro. Cette discipline scientifique est encore relativement récente, mais elle connaît un développement exponentiel. Le défi pour les années à venir sera d’intégrer au vaste fonds des données in vivo les connaissances sur le mécanisme d’action que les études cellulaires et moléculaires auront permis d’accumuler. On pourra ainsi mieux définir les mécanismes de la toxicité et établir un paradigme qui permettra de tirer parti des données in vitro pour prévoir la toxicité in vivo. C’est seulement grâce aux efforts conjugués des toxicologues et des instances gouvernementales que la valeur intrinsèque de ces méthodes in vitro pourra trouver une application concrète.
On entend par étude de la relation structure-activité l’analyse de la structure moléculaire d’un produit chimique pour en tirer des informations prédictives sur ses propriétés essentielles telles que sa stabilité, sa distribution, sa captation, son absorption et sa toxicité. L’étude de la relation structure-activité est l’une des méthodes d’identification des produits chimiques potentiellement dangereux. Elle permet d’isoler les substances exigeant une évaluation complémentaire ou de prendre une décision à un stade précoce pour un produit chimique nouveau et peut, de ce fait, répondre aux attentes de l’industrie et des pouvoirs publics. Les études toxicologiques sont de plus en plus onéreuses et nécessitent des moyens sans cesse plus importants. Préoccupés par le potentiel toxique des produits chimiques auxquels les populations humaines sont exposées, les organismes réglementaires et sanitaires ont cherché à étendre la gamme et la sensibilité des tests pour mettre en évidence le risque toxique. Parallèlement, le poids, réel ou vécu, de la réglementation imposée à l’industrie a amené à s’interroger sur la faisabilité des méthodes d’étude et de l’analyse des données en toxicologie. Actuellement, une étude de cancérogenèse chimique nécessite une expérimentation sur au moins deux espèces, durant leur vie entière, sur les deux sexes, à plusieurs niveaux de doses, à laquelle il faut ajouter l’analyse histopathologique soigneuse de multiples organes sans oublier la détection des lésions prénéoplasiques au niveau des cellules et des organes cibles. Aux Etats-Unis, on estime que les tests de cancérogenèse coûtent plus de 3 millions de dollars (valeur 1995).
Même avec des ressources financières illimitées, tous les toxicologues compétents dans le monde ne seraient pas assez nombreux pour tester les quelque 70 000 produits chimiques existant aujourd’hui. Il faudrait des siècles pour achever ne serait-ce que la première étape de l’évaluation de ces agents chimiques (NRC, 1984). Dans de nombreux pays, l’emploi des animaux pour les expériences de laboratoire a donné lieu à une vague de préoccupations éthiques et à de nouvelles pressions en faveur de l’utilisation des méthodes classiques en expérimentation toxicologique. L’industrie pharmaceutique s’est beaucoup servie des études de la relation structure-activité pour identifier les molécules ayant un potentiel thérapeutique (Hansch et Zhang, 1993). Dans le domaine de l’environnement et de la santé au travail, cette méthode est utilisée pour prévoir la dispersion des produits chimiques dans l’environnement physico-chimique et rechercher les produits chimiques nouveaux ou anciens nécessitant une étude plus approfondie. Aux termes de la loi américaine sur le contrôle des substances toxiques (TSCA), l’EPA applique depuis 1979 la méthode structure-activité comme premier moyen de «dépistage» des nouveaux produits chimiques dans les avis de préfabrication (Premanufacture Notification (PMN)); l’Australie adopte une démarche semblable avec sa procédure NICNAS de déclaration des nouveaux produits chimiques. Aux Etats-Unis, l’étude de la relation structure-activité constitue une étape importante du processus permettant de décider si la fabrication, le traitement, la distribution, l’usage ou la destruction d’une substance présentent un risque excessif pour la santé humaine ou l’environnement, comme le prévoit l’article 5 f) de la TSCA. Sur la base de ses conclusions, l’EPA peut alors exiger, aux termes de l’article 6 du texte de loi précité, que des tests complets de la substance en cause soient effectués.
Sur le plan scientifique, la théorie de la relation structure-activité repose sur le principe selon lequel la structure moléculaire d’un produit chimique permet de prévoir d’importants aspects de son comportement dans les systèmes physico-chimiques et biologiques (Hansch et Leo, 1979).
L’étude de la relation structure-activité comporte les étapes suivantes: identification de la structure chimique, depuis la formule empirique jusqu’au produit pur; identification des analogues structuraux; recherche d’informations dans des bases de données et dans la littérature sur les analogues structuraux; étude de la toxicité et des autres données concernant les analogues structuraux. Il arrive — bien que ce soit assez rare — que l’information sur la structure du produit suffise à elle seule pour justifier la tenue d’une telle étude compte tenu de la bonne compréhension qu’on a des mécanismes de toxicité. Il existe plusieurs bases de données sur les relations structure-activité, de même que des méthodes informatisées de prévision de la structure moléculaire.
A partir de ces informations, la relation structure-activité permet d’évaluer:
Il est à remarquer que la relation structure-activité ne permet pas d’identifier des éléments pourtant importants sur le plan de la santé comme la cancérogénicité, la toxicité du développement, la toxicité de la reproduction, la neurotoxicité, l’immunotoxicité, etc., et ce pour trois raisons: l’absence de bases de données importantes permettant de vérifier les hypothèses sur la relation structure-activité, le manque de connaissances sur les facteurs de nature structurelle induisant une action toxique et la multiplicité des cellules cibles et des mécanismes participant à ces phénomènes (voir l’article «L’approche américaine de l’évaluation du risque des toxiques pour la reproduction et des agents neurotoxiques»). Quelques tentatives limitées d’utilisation de cette méthode ont été faites pour prévoir la pharmacocinétique en se basant sur les coefficients de partage et la solubilité (Johanson et Naslund, 1988). Une étude structure-activité quantitative plus détaillée a été effectuée pour prévoir le métabolisme P450-dépendant d’une série de produits et la liaison, au récepteur cytosolique de la dioxine, de molécules de type dioxine ou PCB (Hansch et Zhang, 1993).
La relation structure-activité permet de prédire — pas toujours de manière très sûre — certains des paramètres énumérés ci-dessus, comme le montre le tableau 33.10. Ce tableau compare les résultats que l’on escompte tirer d’une étude structure-activité à ceux effectivement obtenus grâce à des méthodes expérimentales ou toxicologiques. Des experts de l’EPA ont montré que les études de la relation structure-activité conduites sont plus utiles pour prévoir certaines activités biologiques, en particulier la biodégradation, que pour les propriétés physico-chimiques. S’agissant des différents types de toxicité, la relation structure-activité permet une meilleure prévision de la mutagénicité. Ashby et Tennant (1991), dans une étude plus exhaustive réalisée dans le cadre du programme NTP, montrent également qu’elle garantit une bonne prévisibilité de la génotoxicité à court terme. Ces constatations n’ont rien de surprenant, compte tenu de nos connaissances actuelles sur les mécanismes moléculaires de la génotoxicité (voir l’article «La toxicologie génétique») et le rôle de l’électrophilie sur la liaison à l’ADN. En revanche, ils font remarquer que la relation structure-activité tend à sous-estimer la toxicité systémique et subchronique chez les mammifères et à surestimer la toxicité aiguë pour les organismes aquatiques.
|
Paramètre |
Concordance (%) |
Discordance (%) |
Nombre |
|
Point d�ébullition |
50 |
50 |
30 |
|
Pression de vapeur |
63 |
37 |
113 |
|
Solubilité dans l�eau |
68 |
32 |
133 |
|
Coefficient de partage |
61 |
39 |
82 |
|
Biodégradation |
93 |
7 |
107 |
|
Toxicité sur les poissons |
77 |
22 |
130 |
|
Toxicité sur Daphnia |
67 |
33 |
127 |
|
Toxicité aiguë chez les mammifères (DL50 ) |
80 |
201 |
142 |
|
Irritation cutanée |
82 |
18 |
144 |
|
Irritation oculaire |
78 |
22 |
144 |
|
Sensibilisation cutanée |
84 |
16 |
144 |
|
Toxicité subchronique |
57 |
32 |
143 |
|
Mutagénicité 2 |
88 |
12 |
139 |
|
Mutagénicité 3 |
82�944 |
1�10 |
301 |
|
Cancérogénicité 3: étude sur 2 ans |
72�954 |
— |
301 |
1 La relation structure-activité ne permet pas de prévoir la toxicité aiguë pour 12% des produits chimiques testés. 2 Données de l�OCDE fondées sur la concordance entre le test de Ames et la relation structure-activité. 3 Données NTP fondées sur des études de génotoxicité comparées aux prévisions des études de la relation structure-activité pour plusieurs catégories de �produits chimiques de structure d�alarme�. 4 La concordance varie selon la catégorie: concordance la plus forte avec les produits aromatiques aminés et nitrés; concordance la plus faible avec les structures entrant dans la catégorie �divers�.
Source: données de l�OCDE, communication personnelle C. Auer, EPA (Etats-Unis). Dans cette analyse, seuls ont été utilisés les paramètres pour lesquels on disposait de données comparables obtenues par étude de la relation structure-activité et par études expérimentales. Les données NTP sont extraites de Ashby et Tennant, 1991.
Pour les autres types de toxicité, nous l’avons déjà dit, cette méthode a une utilité moins évidente. Les prévisions de toxicité chez les mammifères sont rendues difficiles en raison de l’absence d’études sur la relation structure-activité pour la toxicocinétique des molécules complexes. Néanmoins, quelques tentatives ont été faites afin de proposer des principes de relation structure-activité pour certains types complexes de toxicité chez les mammifères (voir, par exemple, Bernstein, 1984, pour une étude de ce type concernant les toxiques potentiels sur la reproduction mâle). Dans la plupart des cas, les bases de données ne sont pas suffisamment abondantes pour permettre une analyse rigoureuse des prévisions fondées sur la structure.
A ce stade, on peut conclure que l’étude de la relation structure-activité peut surtout servir à l’affectation de ressources à des investigations toxicologiques plus approfondies ou lever, à un stade précoce, des inquiétudes sur un risque potentiel. Ce type d’étude peut être utilisée de façon fiable comme base d’information pour les décisions ultérieures uniquement dans le cas de la mutagénèse. Mais, comme nous l’expliquons par ailleurs dans ce chapitre et dans l’Encyclopédie, la relation structure-activité ne semble pas pouvoir fournir les informations quantitatives nécessaires à l’évaluation d’un risque, quel qu’il soit.
La toxicologie joue un rôle de premier plan dans l’élaboration des réglementations et des politiques en matière de sécurité et de santé au travail. En effet, pour prévenir les accidents du travail et les maladies professionnelles, on est de plus en plus appelé à prendre des décisions en s’appuyant sur des informations obtenues avant toute exposition humaine ou en l’absence d’une telle exposition. Or, ce type d’informations, comme celles qu’on peut tirer d’études épidémiologiques, renseignerait pourtant de manière définitive sur le risque. De plus, les études toxicologiques, telles qu’elles sont décrites dans ce chapitre, fournissent des données précises sur la dose et le type de réponse dans des conditions contrôlées de laboratoire, informations qu’il est souvent difficile d’obtenir dans le milieu de travail. Cependant, elles doivent être soigneusement évaluées pour estimer la probabilité d’effets nocifs chez l’humain, leur nature et la relation quantitative entre les expositions et les effets.
De nombreux pays s’emploient activement, depuis les années quatre-vingt, à développer des méthodes objectives permettant d’utiliser les informations toxicologiques dans les prises de décisions réglementaires. Des méthodes à l’efficacité prouvée, souvent appelées évaluation du risque, ont été proposées et utilisées dans ces pays à la fois par des instances gouvernementales et non gouvernementales. Pour simplifier les choses, on peut dire de l’évaluation du risque qu’il s’agit d’un processus consistant à apprécier les informations toxicologiques et épidémiologiques et les expositions pour identifier et évaluer la probabilité de survenue d’effets nocifs en association avec l’exposition à des substances ou à des conditions dangereuses. L’évaluation du risque peut avoir un caractère qualitatif, et donc préciser la nature d’un effet nocif et donner une estimation de sa probabilité, ou elle peut avoir un caractère quantitatif et spécifier alors le nombre de personnes affectées à un niveau donné d’exposition. Dans de nombreux systèmes réglementaires, l’évaluation du risque est scindée en quatre étapes: identification du risque, description de la nature de l’effet toxique; évaluation de la relation dose-effet, analyse semi-quantitative ou quantitative de la relation entre l’exposition (ou la dose) et la sévérité ou la probabilité de l’effet toxique; évaluation de l’exposition, évaluation de l’information sur la gamme des expositions pouvant se produire sur une population générale ou sur des sous-groupes à l’intérieur d’une population; caractérisation du risque, compilation de toutes les informations ci-dessus et expression du risque attendu dans des conditions d’exposition spécifiques (voir NRC, 1983, pour l’énoncé de ces principes).
Nous allons présenter à titre d’exemples trois démarches d’évaluation du risque.
Il n’est pas possible de recenser de manière exhaustive toutes les méthodes qui ont cours dans ce domaine à travers le monde, et la sélection proposée ici ne saurait être rigide. Notons que l’on a cherché, notamment sous l’impulsion du GATT, à harmoniser les méthodes d’évaluation du risque. Deux initiatives d’harmonisation internationale ont lieu actuellement, l’une dans le cadre du Programme international sur la sécurité des substances chimiques (PISSC) et l’autre dans celui de l’Organisation pour la coopération et le développement économiques (OCDE). Ces organisations assurent aussi la mise à jour des informations sur les démarches nationales en matière d’évaluation du risque.
Comme dans beaucoup d’autres pays, le risque dû aux produits chimiques est réglementé au Japon en fonction de la catégorie des agents (voir tableau 33.11) par un ministère ou un organisme qui varie selon les cas. Pour les produits chimiques industriels en général, la loi de base qui s’applique est la loi relative à l’examen et à la réglementation de la fabrication, etc., des substances chimiques, ou pour citer son titre abrégé, la «loi sur le contrôle des substances chimiques». Les organismes de tutelle sont le ministère du Commerce international et de l’Industrie et le ministère de la Santé et du Bien-être social. La loi sur la sécurité et la santé au travail (qui relève du ministère du Travail) dispose que le pouvoir mutagène des produits chimiques industriels doit être étudié et que l’exposition des travailleurs à ces produits doit être réduite le plus possible grâce à l’encoffrement des installations de production, à l’aménagement de systèmes d’évacuation ou au port d’un équipement de protection, etc. s’il est établi que le produit est effectivement mutagène.
|
Catégorie |
Loi |
Ministère |
|
Aliments et additifs alimentaires |
Loi sur l�hygiène des produits alimentaires |
MSB |
|
Produits pharmaceutiques |
Loi sur les produits pharmaceutiques |
MSB |
|
Stupéfiants |
Loi sur le contrôle des stupéfiants |
MSB |
|
Produits chimiques agricoles |
Loi sur le contrôle des produits chimiques agricoles |
MAFP |
|
Produits chimiques industriels |
Loi sur le contrôle des produits chimiques industriels |
MSB et MCII |
|
Tous produits chimiques à l�exception des substances radioactives |
Loi réglementant les produits ménagers contenant des substances dangereuses |
MSB |
|
Substances radioactives |
Loi sur les substances radioactives |
AST |
Abréviations: MSB: ministère de la Santé et du Bien-être; MAFP: ministère de l�Agriculture, des Forêts et de la Pêche; MCII: ministère du Commerce international et de l�Industrie; MDT: ministère du Travail; AST: Agence de la science et de la technologie.
Comme l’identification des produits chimiques industriels dangereux relève, pour l’essentiel, de la loi sur le contrôle des substances chimiques, nous décrirons dans le présent article la série de tests prévus à cette fin aux termes de cette loi.
Adoptée à l’origine en 1973 par le parlement japonais (la Diète), entrée en vigueur le 16 avril 1974, la loi sur le contrôle des substances chimiques avait pour principal objectif de prévenir la pollution environnementale et les effets des biphéniles polychlorés (PCB) et substances apparentées sur la santé humaine. Les PCB sont connus pour 1) leur rémanence dans l’environnement (faible biodégradabilité); 2) l’augmentation de leur concentration tout au long de la chaîne alimentaire (bioaccumulation); 3) leur toxicité chronique chez l’humain. En conséquence, la loi impose l’obligation d’étudier ces trois paramètres pour tout produit chimique industriel avant sa commercialisation au Japon. Parallèlement à l’adoption de la loi, la Diète a décidé que l’Agence pour l’environnement devait surveiller le milieu ambiant pour prévenir une éventuelle pollution chimique. La loi a été ensuite modifiée par la Diète en 1986 (modification entrée en vigueur en 1987) pour l’harmoniser avec les décisions de l’OCDE sur la santé et l’environnement, avec l’abaissement des barrières tarifaires en matière de commerce et en particulier avec les directives concernant la mise en place d’une série minimale de données avant commercialisation et les lignes directrices y relatives. Cet amendement prenait en fait acte des conclusions d’une étude de l’environnement réalisée à l’époque et selon laquelle des produits ne s’accumulant pas beaucoup, peu biodégradables et toxiques à long terme, tels que le trichloroéthylène et le tétrachloroéthylène, étaient susceptibles de polluer l’environnement puisqu’on en avait retrouvé la trace dans la nappe phréatique du pays.
La loi classe les produits chimiques industriels en deux catégories: les produits chimiques existants et les nouveaux produits chimiques. Les premiers, répertoriés dans l’«inventaire des produits chimiques existants» (établi lors de l’adoption de la loi d’origine), sont environ au nombre de 20 000, nombre qui dépend de la dénomination qui leur a été donnée dans l’inventaire. Les produits chimiques absents de l’inventaire sont appelés nouveaux produits chimiques. Le gouvernement est chargé de l’identification du risque des produits chimiques existants, tandis que toute compagnie ou tout organisme désirant commercialiser un nouveau produit chimique au Japon est responsable de l’identification du risque pour ce produit. Deux ministères, celui de la Santé et du Bien-être (MSB) et celui du Commerce international et de l’Industrie (MCII) sont chargés de l’application de la loi, l’Agence pour l’environnement pouvant au besoin exprimer son opinion. Les substances radioactives, les toxiques spécifiés, les stimulants et les stupéfiants, réglementés par d’autres lois, ne sont pas visés.
Le schéma d’investigation est représenté à la figure 33.15. Il s’agit, quant au principe, d’un système par étapes qui consiste à étudier la biodégradabilité in vitro de tous les produits chimiques (pour les exceptions, voir ci-après). Si un produit chimique est rapidement biodégradable, il est considéré comme étant «sans danger». Dans le cas contraire, on doit étudier sa bioaccumulation; en cas de «forte accumulation», une étude complète de toxicité est exigée. Sur la base des résultats obtenus, le produit chimique sera classé comme «substance chimique spécifiée de classe 1» lorsque sa toxicité est confirmée, ou alors comme substance «sans danger». Les produits dont l’accumulation est faible ou nulle font l’objet d’études de toxicité, consistant en des tests de mutagenèse et en une administration de doses répétées pendant vingt-huit jours à des animaux (voir tableau 33.12). Après une évaluation complète des données de toxicité, le produit chimique est classé comme «substance chimique désignée» si les résultats mettent en évidence un risque toxique. Sinon, il est considéré comme étant «sans danger». Lorsque d’autres résultats donnent à penser que le produit présente un risque important de pollution environnementale, des données complètes de toxicité sont exigées, à partir desquelles le produit chimique désigné est reclassé comme «substance chimique spécifiée de classe 2» si elles sont positives. Dans le cas contraire, il est considéré comme «sans danger». Le tableau 33.13 indique, outre les grandes lignes de la réglementation, les caractéristiques toxicologiques et écotoxicologiques des différentes catégories de substances: «substance chimique spécifiée de classe 1», «substance chimique spécifiée de classe 2» et «substance chimique désignée».
|
Paramètre |
Plan du test |
|
Biodégradation |
Sur 2 semaines en principe, in vitro, sur des boues activées |
|
Bioaccumulation |
Sur 8 semaines en principe, sur la carpe |
|
Dépistage de la toxicité |
|
|
Administration à doses répétées à 28 jours |
Rats, 3 niveaux de dose, plus lot témoin, pour l�établissement du NOEL, |
|
Substance chimique |
Caractéristiques |
Réglementation |
|
Substances chimiques spécifiées de classe 1 |
Aucune biodégradabilité |
Nécessité d�une autorisation pour la fabrication ou l�importation 1 |
|
Substances chimiques spécifiées de classe 2 |
Aucune biodégradabilité |
Déclaration sur la quantité fabriquée ou importée prévue |
|
Substances chimiques désignées |
Aucune biodégradabilité |
Rapport sur la fabrication ou la quantité importée |
1 En pratique, aucune autorisation.
Au Japon, la loi n’exige pas d’effectuer une étude dans le cas d’un nouveau produit chimique dont la quantité utilisée ne dépasse pas un certain niveau (moins de 1 000 kg/compagnie/an et moins de 1 000 kg/an pour l’ensemble du pays). Les polymères sont examinés selon le schéma des produits de haut poids moléculaire, schéma reposant sur le principe selon lequel les risques d’absorption dans l’organisme sont faibles lorsque le produit chimique a un poids moléculaire supérieur à 1 000 et demeure stable dans l’environnement.
Au cours des vingt-six années qui ont suivi l’entrée en vigueur de la loi sur le contrôle des substances chimiques, soit de 1973 à la fin de 1996, 1 087 produits chimiques existants ont été examinés aux termes de la loi originale puis modifiée. Parmi ces produits, 9 (certains sont identifiés par leur nom générique) ont été classés comme «substance chimique spécifiée de classe 1». Parmi les autres, 36 ont d’abord été placés dans la catégorie des agents «désignés», 23 d’entre eux ont été reclassés comme «substance chimique spécifiée de classe 2» et les 13 autres sont restés «désignés». La figure 33.16 recense les noms des produits chimiques spécifiés de classe 1 et 2. Il en ressort que la plupart des produits chimiques de classe 1 sont des pesticides organochlorés, en dehors des PCB et de leurs substituts et d’un dérivé de l’étain toxique pour les algues. Une majorité de produits chimiques de classe 2 sont des algicides, à l’exception de trois solvants utilisés largement autrefois.
Pendant cette même période de 1973 à la fin 1996, 2 335 nouveaux produits chimiques ont été mis à l’étude, 221 (environ 9,5%) ont été «désignés», mais aucun n’a été rangé dans la catégorie des produits chimiques de classe 1 ou 2. Les autres produits chimiques ont été considérés comme étant «sans danger» et leur fabrication et leur importation ont été autorisées.
Les systèmes nerveux et reproducteur étant très sensibles aux effets des xénobiotiques, la neurotoxicité et la toxicité sur les fonctions de reproduction sont des domaines importants de l’évaluation du risque. De nombreux produits ont été identifiés comme étant toxiques pour ces systèmes chez l’humain (Barlow et Sullivan, 1982; OTA, 1990) ce qui n’est pas surprenant puisque de nombreux pesticides sont délibérément conçus pour perturber la reproduction et la fonction nerveuse dans des organismes cibles, tels que les insectes, en intervenant dans la biochimie hormonale et la neurotransmission.
Il est difficile d’identifier les substances potentiellement toxiques pour ces systèmes pour trois raisons intimement liées: premièrement, ceux-ci font partie des systèmes biologiques les plus complexes chez l’humain, et on estime généralement que les modèles animaux des fonctions reproductrice et neurologique ne représentent pas de manière adéquate les événements critiques tels que la cognition ou le développement précoce embryo-fœtal; deuxièmement, il n’existe pas de tests simples pour identifier les produits potentiellement toxiques pour la reproduction ou le système nerveux; troisièmement, ces systèmes renferment de multiples types cellulaires et de multiples organes, de sorte qu’il n’est pas possible d’envisager un mécanisme de toxicité unique pour en inférer une relation dose-réponse ou prévoir une relation structure-activité. On sait, de plus, que la sensibilité du système nerveux et du système reproducteur varie en fonction de l’âge, et que l’exposition peut avoir des effets plus sévères à certaines périodes critiques qu’à d’autres.
La neurotoxicité est un problème de santé publique important. Comme le montre le tableau 33.14, plusieurs accidents neurotoxiques graves se sont produits qui ont touché des milliers de travailleurs et diverses populations à l’occasion de pollutions industrielles ou de la contamination d’aliments, d’eau ou d’autres vecteurs. On sait aussi que les expositions professionnelles à des neurotoxiques tels que le plomb, le mercure, les insecticides organophosphorés et les solvants chlorés sont largement répandues à travers le monde (OTA, 1990; Johnson, 1978).
|
Année(s) |
Lieu |
Substance |
Observations |
|
400 avant J.-C. |
Rome |
Plomb |
Hippocrate découvre la toxicité du plomb dans l�industrie minière |
|
Années 1930 |
Etats-Unis (sud-est) |
TOCP |
Composé souvent ajouté aux huiles de lubrification: contamination du �Ginger Jake�, boisson alcoolisée, 20 000 à 100 000 cas, dont plus de 5 000 personnes paralysées |
|
Années 1930 |
Europe |
Apiol (avec TOCP) |
Médicament abortif contenant du TOCP: 60 cas de neuropathie |
|
1932 |
Etats-Unis (Californie) |
Thallium |
Vol d�orge traité par du sulfate de thallium, utilisé comme rodenticide, puis utilisation pour la confection de tortillas; hospitalisation de 13 membres d�une famille présentant des symptômes neurologiques; 6 d�entre eux décèdent |
|
1937 |
Afrique du Sud |
TOCP |
Soixante Sud-Africains sont victimes d�une paralysie après avoir employé une huile alimentaire contaminée |
|
1946 |
— |
Tétraéthylplomb |
Plus de 25 individus présentent des troubles neurologiques après nettoyage de réservoirs d�essence |
|
Années 1950 |
Japon (Minimata) |
Mercure |
Ingestion de poissons et de crustacés contaminés par du mercure provenant d�une usine chimique; 121 personnes intoxiquées, 46 morts, nombreux enfants présentant de graves lésions du système nerveux |
|
Années 1950 |
France |
Organostanneux |
Contamination du Stalinon par du triéthylétain responsable de plus de 100 morts |
|
Années 1950 |
Maroc |
Manganèse |
Cent cinquante mineurs présentent une intoxication chronique au manganèse avec troubles neurocomportementaux sévères |
|
Années 1950 à 1970 |
Etats-Unis |
AETT |
Composant de parfums, neurotoxique, retiré du marché en 1978; effets sur la santé humaine non connus |
|
1956 |
— |
Endrine |
Quarante-neuf personnes tombent malades après avoir consommé des produits de boulangerie confectionnés avec de la farine contaminée par un insecticide, l�endrine; quelques cas de convulsions |
|
1956 |
Turquie |
HCB |
Hexachlorobenzène, fongicide de graines de semences, intoxication de 3 000 à 4 000 personnes; 10% d�entre elles décèdent |
|
1956-1977 |
Japon |
Clioquinol |
Médicament utilisé pour traiter la diarrhée des voyageurs: neuropathie ayant touché 10 000 personnes en 20 ans |
|
1959 |
Maroc |
TOCP |
Huile alimentaire contaminée par de l�huile de moteur: environ 10 000 individus atteints |
|
1960 |
Iraq |
Mercure |
Graines de semence traitées par du mercure fongicide et utilisées pour faire du pain; plus de 1 000 personnes atteintes |
|
1964 |
Japon |
Mercure |
Méthylmercure: intoxication de 646 personnes |
|
1968 |
Japon |
PCB |
Biphényles polychlorés dans un produit à base de riz: 1 665 personnes atteintes |
|
1969 |
Japon |
n-Hexane |
Quatre-vingt-treize cas de neuropathie après exposition au n-hexane entrant dans la fabrication de sandales en vinyle |
|
1971 |
Etats-Unis |
Hexachlorophène |
Produit utilisé pendant des années comme désinfectant en solution à 3% pour la toilette des nourrissons: toxique du système nerveux et d�autres systèmes |
|
1971 |
Iraq |
Mercure |
Graines de semence, traitées par un composé mercuriel fongicide et utilisées pour faire du pain: plus de 5 000 cas d�intoxications sévères, 450 morts, effets non documentés sur de nombreux nourrissons exposés en période prénatale |
|
1973 |
Etats-Unis (Ohio) |
MIBK |
Employés d�une usine textile exposés à ce solvant; plus de 80 travailleurs atteints de neuropathie, 180 présentant des atteintes moins graves |
|
1974-1975 |
Etats-Unis (Hopewell, Virginie) |
Chlordécone (Képone) |
Employés d�une usine chimique exposés à cet insecticide; plus de 20 travailleurs présentent des problèmes neurologiques sévères, plus de 40 des troubles moins graves |
|
1976 |
Etats-Unis (Texas) |
Leptophos (Phosvel) |
Neuf employés au moins souffrent de problèmes neurologiques sévères par suite de l�exposition lors de la fabrication |
|
1977 |
Etats-Unis (Californie) |
Dichloropropène |
Vingt-quatre sujets hospitalisés après exposition au pesticide Télone à la suite d�un accident de circulation |
|
1979-1980 |
Etats-Unis (Lancaster, Texas) |
BHMH (Lucel-7) |
Sept employés d�une fabrique de douches en plastique présentent des troubles neurologiques après une exposition au BHMH |
|
Années 1980 |
Etats-Unis |
MPTP |
Impureté de synthèse d�une drogue illicite provoquant des symptômes identiques à ceux de la maladie de Parkinson |
|
1981 |
Espagne |
Huile toxique contaminée |
Quelque 20 000 personnes intoxiquées par une substance toxique dans de l�huile: plus de 500 morts; de nombreuses personnes présentent une neuropathie grave |
|
1985 |
Etats-Unis et Canada |
Aldicarb |
Plus de 1 000 personnes en Californie et dans d�autres Etats de l�ouest et en Colombie-Britannique présentent des problèmes neuromusculaires et cardiaques par suite de l�ingestion de melons contaminés par ce pesticide |
|
1987 |
Canada |
Acide domoïque |
Ingestion de moules contaminées par de l�acide domoïque: 129 malades et 2 morts; symptômes: perte de mémoire, désorientation et convulsions |
Source: OTA, 1990.
Les produits chimiques peuvent atteindre l’une des nombreuses cibles cellulaires ou biochimiques présentes dans le système nerveux central ou périphérique. Des effets toxiques s’exerçant sur d’autres organes peuvent également affecter le système nerveux, comme le montre l’exemple de l’encéphalopathie hépatique. La neurotoxicité se manifeste par des effets sur les processus d’apprentissage (incluant mémoire, faculté cognitive et performance intellectuelle), les processus somato-sensoriels (dont la sensibilité et la proprioception), la fonction motrice (équilibre, démarche, contrôle fin des mouvements, etc.), l’affect (notamment la personnalité et l’émotivité) et la fonction autonome (contrôle nerveux de la fonction endocrine et des organes internes). Les effets toxiques des produits chimiques sur le système nerveux sont plus ou moins intenses et se manifestent de façon différente selon l’âge du sujet: au cours du développement, le système nerveux central est particulièrement vulnérable en raison du processus de la différenciation cellulaire, de la migration et du contact intercellulaire qui prend place chez l’humain (OTA, 1990). De plus, les lésions cytotoxiques sur le système nerveux sont souvent irréversibles, les neurones n’étant pas remplacés après l’embryogenèse. Bien que le système nerveux central (SNC) soit relativement protégé du contact avec les produits absorbés grâce à un rempart très étanche (la barrière hémato-encéphalique, formée de cellules capillaires endothéliales qui tapissent le système vasculaire cérébral), les produits chimiques toxiques sont néanmoins susceptibles d’atteindre le SNC par trois mécanismes: les solvants et les composés lipophiles peuvent passer à travers les membranes cellulaires; certains produits peuvent se fixer aux protéines de transport endogènes qui fournissent des nutriments et des molécules biologiques au SNC; enfin, de petites molécules, si elles sont inhalées, vont être directement absorbées par le nerf olfactif et transportées au cerveau.
Aux Etats-Unis, quatre organismes ont le pouvoir de réglementer les substances neurotoxiques: la FDA, l’EPA, l’OSHA et la CPSC. L’OSHA réglemente généralement les expositions professionnelles aux produits chimiques neurotoxiques (entre autres), l’EPA ayant compétence pour les expositions professionnelles et non professionnelles aux pesticides, aux termes de la loi fédérale sur les insecticides, fongicides et rodenticides (Federal Insecticide, Fungicide and Rodenticide Act (FIFRA)), mais aussi pour les nouveaux produits chimiques. C’est elle qui intervient avant leur fabrication et leur commercialisation et, à ce titre, elle doit tenir compte à la fois des risques professionnels et non professionnels.
Les agents qui ont un effet nocif sur la physiologie, la biochimie ou l’intégrité structurale du système nerveux ou son fonctionnement au niveau comportemental sont considérés comme des produits présentant un risque neurotoxique (EPA, 1993). Il n’est pas facile d’établir le pouvoir neurotoxique d’un produit, en raison notamment de la complexité du système nerveux et des multiples expressions de la neurotoxicité. Certains effets peuvent paraître retardés, comme la neurotoxicité différée de certains insecticides organophosphorés. Il faut donc faire montre de prudence et de discernement lorsqu’on doit se prononcer sur un tel risque et tenir compte des conditions de l’exposition, de la dose, de la durée et du rythme.
L’identification du risque se fait généralement à partir d’études toxicologiques sur des organismes intacts, chez lesquels les fonctions comportementale, cognitive, somato-motrice et somato-sensorielle sont évaluées grâce à toute une panoplie d’outils d’investigation dont la biochimie, l’électrophysiologie et la morphologie (Tilson et Cabe, 1978; Spencer et Schaumberg, 1980). On ne saurait assez insister sur l’importance de l’observation attentive du comportement de l’organisme entier. La toxicité doit aussi être jaugée à différentes étapes du développement, y compris aux premiers stades de la vie (intra-utérine et néonatale) et à celui de la sénescence. Chez l’être humain, la mise en évidence de la neurotoxicité suppose qu’on effectue une appréciation clinique grâce à des méthodes d’évaluation neurologique de la fonction motrice, du langage, des réflexes, de la fonction sensorielle, de l’électrophysiologie, de l’étude neuropsychologique et, dans certains cas, grâce à des techniques avancées d’imagerie du cerveau et d’électroencéphalographie quantitative. L’OMS a mis au point une batterie de tests neurocomportementaux, incluant des témoins de la fonction motrice, de la coordination main-œil, du temps de réaction, de la mémoire immédiate, de l’attention et de l’humeur. Cette batterie a été validée au niveau international dans le cadre d’une initiative coordonnée (Johnson, 1978).
Chez les animaux aussi, l’identification du risque doit se faire au moyen de méthodes d’observation minutieuses. L’EPA a développé une batterie d’observations fonctionnelles qui constitue une première étape à la fois pour déceler et quantifier les effets neurotoxiques majeurs patents (Moser, 1990). Ces paramètres sont inclus dans les tests de toxicité subchronique et chronique de l’OCDE. Une batterie typique porte sur les paramètres suivants: posture; démarche; mobilité; fonction d’éveil et réactivité; présence ou absence de tremblements, convulsions, larmes, horripilation, salivation, miction ou défécation excessives, stéréotypie, mouvements rotatoires ou autres comportements anormaux. Les comportements provoqués incluent la réponse à la manipulation, au pincement de la queue, ou aux claquements; équilibre, réflexe de redressement et force d’agrippement du membre postérieur. Le tableau 33.15 dresse la liste d’un certain nombre de tests représentatifs accompagnée d’exemples de produits qu’ils permettent d’identifier.
|
Fonction |
Procédé |
Agents responsables |
|
Neuromusculaire |
||
|
Faiblesse |
Force de préhension; endurance à la nage; suspension à une barre; fonction motrice discriminante; écartement des membres postérieurs |
n-Hexane, méthylbutylcétone, carbaryle |
|
Incoordination |
Rotarod, aspect de la démarche |
3-Acétylpyridine, éthanol |
|
Tremblement |
Echelle d�évaluation, analyse spectrale |
Chlordécone, pyréthroïdes de type 1, DDT |
|
Myoclonie, spasmes |
Echelle d�évaluation, analyse spectrale |
DDT, pyréthroïdes de type 2 |
|
Sensorielle |
||
|
Audition |
Conditionnement discriminant, modification de réflexe |
Toluène, triméthylétain |
|
Toxicité visuelle |
Conditionnement discriminant |
Méthylmercure |
|
Toxicité somatosensorielle |
Conditionnement discriminant |
Acrylamide |
|
Sensibilité à la douleur |
Conditionnement discriminant; batterie d�observation fonctionnelle |
Parathion |
|
Toxicité olfactive |
Conditionnement discriminant |
3-Méthylindole méthylbromure |
|
Apprentissage, mémoire |
||
|
Accoutumance |
Réflexe de sursaut |
Diisopropylfluorophosphate (DFP) |
|
Conditionnement classique |
Membrane nictitante, aversion du goût conditionné, évitement passif, conditionnement olfactif |
Aluminium, carbaryle, triméthylétain, IDPN, triméthylétain (néonatal) |
|
Conditionnement opérant ou instrumental |
Evitement une voie, évitement deux voies, évitement de labyrinthe Y, labyrinthe d�eau Biol, labyrinthe d�eau Morris, labyrinthe de bras radial, appariement différé à l�échantillon, acquisition répétée, apprentissage de discrimination visuelle |
Chlordécone, plomb (néonatal), hypervitaminose A, styrène, DFP, triméthylétain, DFP, carbaryle, plomb |
Source: EPA, 1993.
Une fois ces tests effectués, on peut procéder à des évaluations plus complexes, réservées en général aux études de mécanisme plutôt que d’identification du risque. Les méthodes in vitro pour l’identification du risque neurotoxique sont d’une application limitée, car elles ne renseignent aucunement sur les effets au niveau des fonctions complexes, telles que l’apprentissage. Par contre, elles peuvent être très utiles pour définir les cibles de la toxicité et améliorer la précision des études dose-réponse au niveau des cibles (voir OMS, 1986 et EPA, 1993 pour des observations détaillées des principes et des méthodes d’identification des neurotoxiques potentiels).
Nous avons expliqué que la relation entre la toxicité et la dose peut être établie à partir de données humaines, quand elles existent, ou à partir d’études sur animaux. Aux Etats-Unis, on emploie en général pour les neurotoxiques une approche de type facteur d’incertitude ou de sécurité. Pour cela, on fixe un «niveau sans effet nocif observé» (No Observed Adverse Effect Level (NOAEL)) ou le «niveau du plus faible effet nocif observé» (Lowest Observed Adverse Effect Level (LOAEL)), le résultat obtenu étant ensuite divisé par un facteur d’incertitude ou de sécurité (généralement un multiple de 10), qui permet de tenir compte de l’imperfection des données, de la sensibilité potentielle plus élevée chez l’humain et de la variabilité de la réponse humaine en fonction de l’âge ou d’autres paramètres. Le nombre qui résulte de ce calcul est appelé dose de référence (DRf) ou concentration de référence (CRf). Pour établir le LOAEL ou le NOAEL, on se fonde en général sur l’effet produit à la dose la plus faible chez l’espèce animale et le sexe les plus sensibles. La conversion de la dose animale à l’exposition humaine est effectuée grâce à des méthodes normalisées de dosimétrie interespèces qui tiennent compte des différences de durée de vie et de durée d’exposition.
L’utilisation d’un facteur de sécurité suppose qu’il existe un seuil ou une dose au-dessous desquels aucun effet nocif ne se produit. Il peut s’avérer difficile d’établir des seuils pour des neurotoxiques spécifiques par la voie expérimentale; ces seuils sont basés sur des hypothèses relatives au mécanisme d’action qui peuvent ne pas convenir pour tous les neurotoxiques (Silbergeld, 1990).
A ce stade, les sources, les voies, les doses et les temps d’exposition à un neurotoxique sont évaluées dans une population humaine, une sous-population ou même chez des individus. Cette information peut être obtenue grâce à des contrôles d’ambiance ou à partir d’échantillons humains, par des estimations basées sur des conditions normalisées d’exposition (telles que les conditions du lieu de travail et les tâches effectuées) ou encore à partir de modèles sur le devenir et la dispersion dans l’environnement (voir EPA, 1992, pour les directives générales relatives aux méthodes d’évaluation de l’exposition). Dans quelques cas limités, on peut recourir aux indicateurs biologiques pour valider les conclusions et les estimations sur l’exposition, bien que les indicateurs utilisables pour les neurotoxiques soient assez peu nombreux.
Pour caractériser le risque, on doit l’identifier et évaluer la relation dose-réponse et l’exposition. Pour cela, on est appelé à faire des hypothèses sur l’extrapolation des fortes doses aux faibles doses ou de l’animal à l’humain, ainsi que sur l’adéquation des hypothèses de seuil et des facteurs de sécurité.
Les risques pour la reproduction peuvent concerner de multiples paramètres fonctionnels et cibles cellulaires chez l’humain et se traduire par des effets au niveau du sujet atteint et de sa descendance. Ces risques peuvent affecter le développement du système reproducteur chez le mâle ou la femelle, les comportements sexuels, la fonction hormonale, l’hypothalamus et l’hypophyse, les gonades et les cellules germinales, la fertilité, la gestation et la durée de la fonction de reproduction (OTA, 1985). De plus, les produits chimiques mutagènes peuvent également affecter la fonction reproductrice en lésant l’intégrité des cellules germinales (Dixon, 1985).
La nature et l’étendue des effets nocifs d’une exposition chimique sur les fonctions de reproduction dans les populations humaines sont encore très peu connues. On dispose d’assez peu d’informations sur la surveillance de paramètres tels que la fertilité de l’homme ou de la femme, l’âge de la ménopause chez la femme, ou la numération spermatique chez l’homme. Pourtant, des hommes et des femmes sont employés dans des branches où ils sont exposés à des produits présentant un danger pour la fonction de reproduction (OTA, 1985).
La présente rubrique met l’accent sur les problèmes spécifiques à l’évaluation du risque des toxiques pour la reproduction, sans revenir sur les aspects communs à l’évaluation des risques touchant à la fois le système reproducteur et le système nerveux. Comme pour les neurotoxiques, la responsabilité en matière de réglementation des produits affectant la fonction de reproduction incombe à l’EPA, l’OSHA, la FDA et le CPSC. Parmi ces organismes, seule l’EPA s’est dotée de lignes directrices permettant l’évaluation du risque pour les fonctions de reproduction. Aux Etats-Unis, l’Etat de Californie a lui aussi élaboré des méthodes pour évaluer le risque toxique sur les fonctions de reproduction, aux termes d’une loi d’Etat, la proposition 65 (Pease et coll., 1991).
Les agents toxiques pour les fonctions reproductrices, comme les neurotoxiques, peuvent affecter l’un des nombreux organes cibles ou sites moléculaires de cette fonction. Leur évaluation est complexe puisqu’il est nécessaire d’évaluer séparément et simultanément trois organismes distincts: le mâle, la femelle et la descendance (Mattison et Thomford, 1989). Alors que l’un des critères essentiels de la fonction reproductrice est la conception d’un enfant sain, la biologie de la reproduction intervient également dans la santé des organismes en développement ou matures indépendamment de leur implication dans la procréation. Par exemple, la perte de la fonction ovulatoire par déplétion naturelle ou ablation chirurgicale des ovaires influe sur la santé des femmes, puisqu’elle entraîne des modifications de la pression sanguine, du métabolisme lipidique et de la physiologie osseuse. Les modifications biochimiques hormonales peuvent être à l’origine d’une plus grande prédisposition au cancer.
L’identification d’un risque sur les fonctions de reproduction peut se faire à partir de données humaines ou animales. En général, les données humaines sont assez peu nombreuses, car elles ne peuvent être obtenues qu’au prix d’une surveillance astreignante pour détecter les troubles de la fonction reproductrice, tels que le spermogramme, la fréquence ovulatoire et la longueur du cycle, ou l’âge de la puberté. La détection d’un risque sur la reproduction à partir d’informations sur les taux de fertilité ou de données sur le nombre de grossesses peut être bouleversée par la suppression intentionnelle de fertilité exercée par beaucoup de couples dans le cadre des mesures de planification familiale. La surveillance minutieuse de certaines populations montre que le taux d’échec de la reproduction (avortements) peut être très élevé, lorsqu’on évalue des indicateurs biologiques de début de grossesse (Sweeney et coll., 1988).
Les protocoles d’étude faisant appel aux animaux d’expérience sont largement employés pour identifier les toxiques agissant sur la reproduction. Dans la plupart de ces études, comme celles effectuées aux Etats-Unis par la FDA et l’EPA et, au niveau international, selon les directives de l’OCDE, les effets d’agents suspects sont mis en évidence d’après: la fertilité après exposition du mâle ou de la femelle; l’observation des comportements sexuels au moment de l’accouplement; et l’examen histopathologique des gonades et des glandes sexuelles accessoires, telles que les glandes mammaires (EPA, 1994). Les études de toxicité sur les fonctions de reproduction impliquent souvent l’administration du produit aux animaux sur une ou plusieurs générations afin de déceler les effets sur le processus de la reproduction dans son intégralité et d’étudier les effets sur les organes spécifiques de la reproduction. Il est recommandé de faire porter les études sur plusieurs générations, car elles permettent de détecter des effets induits par une exposition lors du développement du système reproducteur in utero. Un protocole spécial, le protocole d’évaluation de la toxicité sur la reproduction par administration continue (RACB), a été mis au point aux Etats-Unis dans le cadre du Programme national de toxicologie. Ce test fournit des données sur les modifications de l’espacement temporel des parturitions (reflétant la fonction ovulatoire) et sur le nombre et la taille des portées pendant toute la période du test. Quand le test est effectué tout au long de la vie de la femelle, il donne des informations sur les premiers troubles de reproduction. Les mesures sur le sperme peuvent être ajoutées au RACB pour déceler les modifications de la fonction reproductrice chez le mâle. Le test du dominant létal, qui est un test spécial pour détecter la perte de pré- ou postimplantation, contribue à mettre en évidence les effets mutagènes sur la spermatogenèse.
Des tests in vitro ont également été mis au point comme tests de dépistage de la toxicité sur les fonctions de reproduction (et sur le développement) (Heindell et Chapin, 1993). Ces tests, qui sont généralement utilisés en complément des tests in vivo, fournissent davantage d’informations sur la cible et sur le mécanisme des effets observés.
Le tableau 33.16 montre les trois types de paramètres d’évaluation de la toxicité sur les fonctions de reproduction représentatifs du couple ou spécifiques de la femelle ou du mâle. Les paramètres représentatifs du couple incluent ceux qui sont étudiés sur plusieurs générations et sur l’organisme seul. Ils incluent également en général l’évaluation de la portée. A noter que la mesure de la fertilité chez les rongeurs manque généralement de sensibilité, par rapport aux mesures effectuées chez l’humain et que les effets nocifs sur la fonction reproductrice peuvent se produire à de bien plus faibles doses que celles qui affectent la fertilité de manière significative (EPA, 1994). Les paramètres spécifiques du mâle peuvent inclure les tests du dominant létal de même que l’évaluation histopathologique des organes et du sperme, le dosage des hormones et des marqueurs du développement sexuel. La fonctionnalité du sperme peut également être évaluée par des méthodes de fertilisation in vitro pour détecter les propriétés de pénétration et les aptitudes des cellules germinales; ces tests sont précieux parce qu’ils sont directement comparables aux évaluations in vitro conduites dans les centres de fertilité humaine, même s’ils ne fournissent pas par eux-mêmes une information dose-réponse. Les paramètres propres à la femelle incluent, en plus de l’histopathologie des organes et des dosages hormonaux, l’évaluation des suites de la reproduction, dont la lactation et la croissance de la portée.
|
|
Paramètres liés au couple |
|
Etudes multigénérations |
Autres paramètres de la reproduction |
|
Taux et durée d�accouplement (temps de gestation1) |
Taux d�ovulation |
|
Taux de gestation1 |
Taux de fécondation |
|
Taux de délivrance1 |
Perte avant implantation |
|
Durée de gestation1 |
Nombre d�implantations |
|
Taille de la portée (totale et vivante) |
Perte après implantation1 |
|
Nombre de petits vivants et morts (taux de mortalité fœtale)1 |
Malformations et modifications internes1 |
|
Sexe de la progéniture |
Développement structurel et fonctionnel postnatal1 |
|
Poids à la naissance1 |
|
|
Poids postnatal1 |
|
|
Survie de la progéniture1 |
|
|
Malformations et modifications externes1 |
|
|
Reproduction de la progéniture1 |
|
|
|
Paramètres spécifiques du mâle |
|
Poids des organes |
Testicules, épididyme, vésicules séminales, prostate, hypophyse |
|
Examen visuel et histopathologie |
Testicules, épididyme, vésicules séminales, prostate, hypophyse |
|
Evaluation du sperme1 |
Numération spermatique et qualité (morphologie, motilité) |
|
Taux hormonaux1 |
Hormone lutéinisante, FSH, testostérone, œstrogène, prolactine |
|
Développement |
Descente des testicules1, séparation préputiale, production de sperme1, distance ano-génitale, aspect des organes génitaux externes1 |
|
|
Paramètres spécifiques de la femelle |
|
Poids corporel |
|
|
Poids des organes |
Ovaire, utérus, vagin, hypophyse |
|
Examen visuel et histopathologie |
Ovaire, utérus, vagin, hypophyse, oviducte, glande mammaire |
|
Normalité du cycle œstral (menstruel1) |
Cytologie du frottis vaginal |
|
Taux hormonaux1 |
LH, FSH, œstrogène, progestérone, prolactine |
|
Lactation1 |
Croissance de la progéniture |
|
Développement |
Normalité des organes génitaux externes1, ouverture vaginale, cytologie du frottis vaginal, début du fonctionnement de l�œstrus (menstruation1) |
|
Sénescence (ménopause1) |
Cytologie du frottis vaginal, histologie ovarienne |
1 Paramètres pouvant être obtenus de manière relativement non invasive chez l�humain.
Source: EPA, 1994.
Aux Etats-Unis, l’identification du risque se termine par une évaluation qualitative des données sur la toxicité qui permet de classer les produits chimiques selon qu’ils présentent ou non une preuve suffisante de risque (EPA, 1994). Par preuve «suffisante», on entend des données épidémiologiques fournissant des arguments convaincants de la relation causale (ou de son absence), basés sur des études cas-témoins ou de cohortes, ou des séries de cas bien étayés. Des données suffisantes sur les animaux peuvent être couplées à des données limitées chez l’humain pour confirmer l’existence d’un risque sur la reproduction: pour être suffisantes, les études expérimentales doivent en général appliquer les directives de l’EPA sur deux générations et comporter un minimum de données démontrant un effet nocif sur la fonction reproductrice dans une étude bien conduite sur une espèce appropriée. Des données humaines limitées peuvent être disponibles ou non; elles ne sont pas nécessaires aux fins de l’identification du risque. Pour exclure un risque potentiel sur la reproduction, les données animales doivent inclure un ensemble de paramètres établi à partir de plusieurs études montrant l’absence d’effet nocif sur la reproduction aux doses minimales toxiques pour l’animal (EPA, 1994).
Comme dans le cas de l’évaluation des agents neurotoxiques, la démonstration d’effets liés à la dose constitue une partie importante de l’évaluation du risque pour les toxiques de la reproduction. Les analyses dose-réponse achoppent sur deux difficultés: la toxicocinétique complexe lors de la gestation, et l’importance de distinguer la toxicité qui affecte véritablement la reproduction de la toxicité générale sur l’organisme. Les animaux affaiblis, ou ceux présentant une toxicité non spécifique notable (une perte de poids par exemple) peuvent ne pas ovuler ou s’accoupler. Une toxicité maternelle peut affecter la viabilité de la gestation ou la possibilité de lactation. Ces effets, bien que révélateurs d’une toxicité, ne sont pas spécifiques de la reproduction (Kimmel et coll., 1986). L’évaluation de la relation dose-réponse pour un paramètre spécifique, tel que la fertilité, doit s’effectuer dans le cadre d’une étude générale de la reproduction et du développement. Les relations dose-réponse pour différents effets peuvent être individuellement significatives, mais interférer entre elles. Ainsi, des agents qui réduisent la taille de la portée peuvent ne pas avoir d’effet sur le poids de celle-ci en raison d’une faible interférence sur la nutrition intra-utérine.
Le moment et la durée des expositions sont des facteurs qui revêtent une grande importance lorsqu’on se propose d’évaluer la toxicité sur la reproduction. Selon le processus biologique en cause, les mesures cumulatives de l’exposition peuvent ne pas être suffisamment précises. On sait que l’exposition peut avoir des conséquences variables aussi bien chez l’humain que chez l’animal selon le stade de développement auquel elle se produit chez le mâle et chez la femelle (Gray et coll., 1988). Les conséquences d’un effet toxique au niveau de la spermatogenèse ou de l’ovulation dépendent également du moment où elles se produisent. Les effets sur la spermatogenèse peuvent être réversibles si l’exposition cesse, alors que la toxicité sur l’ovocyte n’est pas réversible puisque le nombre de cellules germinales utilisables pour l’ovulation est fixe (Mattison et Thomford, 1989).
Comme pour les neurotoxiques, on suppose qu’il existe de façon générale un seuil pour les toxiques de la reproduction. Cependant, l’action des agents mutagènes sur les cellules germinales peut être considérée comme une exception à cette hypothèse générale. Pour les autres paramètres, on calcule une DRf (dose de référence) ou une CRf (concentration de référence) comme pour les neurotoxiques en déterminant le NOAEL ou le LOAEL et en appliquant un facteur de sécurité. L’effet utilisé pour établir le NOAEL ou le LOAEL est le paramètre le plus sensible chez l’espèce mammifère la plus appropriée et la plus sensible (EPA, 1994). Le facteur de sécurité prend en compte les variations interespèces et intraespèce, la possibilité de définir un véritable NOAEL ainsi que la sensibilité du paramètre détecté.
La caractérisation du risque devrait également s’intéresser à des groupes spécifiques de population soumis au risque, en tenant notamment compte des différences mâle-femelle, de l’état de la gestation et de l’âge. On devra prendre en considération les sujets particulièrement sensibles, comme les femmes allaitant, celles dont le nombre d’ovocytes est réduit, les hommes à faible numération spermatique, ou encore les adolescents prépubères.
Depuis 1971, le Centre international de recherche sur le cancer (CIRC) publie, dans le cadre de son programme d’identification du risque cancérogène chez l’être humain, une série de monographies sous le titre Monographs on the Evaluation of Carcinogenic Risks to Humans (Monographies du CIRC sur l’évaluation des risques de cancérogénicité pour l’homme). A ce jour, 73 volumes ont paru ou sont sous presse et 833 agents ou circonstances d’exposition ont fait l’objet d’une étude de cancérogénicité (voir annexe).
Ces évaluations qualitatives du risque cancérogène chez l’humain correspondent à la phase d’identification dans le schéma généralement admis d’évaluation du risque qui comprend, outre l’identification du risque à proprement parler, l’évaluation de la relation dose-réponse (avec une extrapolation à des valeurs pour lesquelles on ne dispose pas de données), l’évaluation de l’exposition et la caractérisation du risque en question.
Le programme du CIRC a pour but de procéder — avec l’aide de groupes de travail composés d’experts internationaux — à l’examen critique et à l’évaluation des données concernant le pouvoir cancérogène d’agents (produits chimiques, groupes de produits chimiques, mélanges, facteurs physiques ou biologiques) ou de circonstances d’exposition (expositions professionnelles, habitudes culturelles), puis de les publier. Les groupes de travail préparent des monographies sur les agents individuels ou les expositions et chaque volume est publié et diffusé le plus largement possible. Chaque monographie contient une rubrique sur les propriétés physico-chimiques de l’agent, les méthodes d’analyse, le mode de production, la quantité produite, l’utilisation, les conditions d’exposition et l’exposition humaine, et des résumés de cas isolés et d’études épidémiologiques de cancer humain. On y trouve également des résumés des tests de cancérogenèse expérimentale, une brève description des autres données biologiques importantes, telles que la toxicité et les effets génétiques, qui peuvent orienter sur le mécanisme d’action et, enfin, une évaluation globale du pouvoir cancérogène. La première partie de ce schéma général peut être adaptée lorsqu’il s’agit d’agents autres que des produits chimiques ou de mélanges de produits chimiques.
Les principes directeurs de l’évaluation des agents cancérogènes, qui ont été arrêtés par divers groupes d’experts spéciaux, figurent dans le préambule des monographies (CIRC, 1994a).
Les données obtenues lors d’études réalisées chez des sujets exposés, lors d’expériences sur des animaux de laboratoire ou encore à l’occasion d’études sur l’exposition, le métabolisme, la toxicité et les effets génétiques chez l’humain et les animaux sont dépouillées, après quoi des corrélations sont établies.
Trois types d’études épidémiologiques contribuent à l’évaluation de la cancérogénicité pour l’humain: les études de cohortes, les études cas-témoins et les études de corrélation (ou écologiques). A ces trois types d’études peut s’ajouter l’examen de cas isolés de cancer.
Les études de cohortes et les études cas-témoins associent les différentes expositions étudiées à l’apparition de cancer chez les sujets et donnent une estimation du risque relatif (rapport entre l’incidence ou la mortalité chez les personnes exposées et l’incidence ou la mortalité chez les personnes non exposées) comme principale mesure d’association.
Dans les études de corrélation, les unités d’investigation sont le plus souvent des populations entières (comme par exemple dans des secteurs géographiques particuliers) et la fréquence des cancers est associée à une mesure globale de l’exposition de la population à l’agent, au mélange ou à la circonstance d’exposition étudiés. Comme l’exposition individuelle n’est pas connue, un rapport de cause à effet est cependant moins facile à prouver à partir d’études de corrélation qu’à partir d’études de cohortes ou d’études cas-témoins. Les rapports de cas isolés se basent généralement sur un soupçon, fondé sur l’expérience clinique, de ce que la coïncidence de deux événements — à savoir une exposition particulière et la survenue d’un cancer — a été plus fréquente que ce que l’on aurait pu attendre du seul fait du hasard. Les incertitudes entourant l’interprétation des rapports de cas et des études de corrélation les rendent, sauf exception, insuffisants pour constituer la seule base permettant de conclure à un rapport causal.
Il est nécessaire de prendre en compte le rôle possible de biais et de facteurs de confusion dans l’interprétation des études épidémiologiques. Par «biais», on entend l’intervention, dans le protocole d’une étude ou son déroulement, de facteurs qui mèneraient de façon erronée à surestimer ou à sous-estimer l’association entre la maladie et un agent, un mélange ou une circonstance d’exposition. Par «confusion», on entend une situation dans laquelle la relation avec la maladie apparaît plus étroite ou moins étroite qu’elle n’est réellement en raison d’une association entre le facteur causal apparent et un autre facteur associé à une augmentation ou à une diminution de l’incidence de la maladie.
Dans l’évaluation des études épidémiologiques, une association franche (à savoir un risque relatif élevé) est un meilleur indicateur de causalité qu’une association lâche, bien que l’on sache qu’un risque relatif faible n’implique pas nécessairement une absence de causalité et qu’il peut être important si la maladie est courante. Les associations répétées, dans plusieurs études de protocole identique ou utilisant différentes méthodes épidémiologiques, ou encore menées dans différents contextes d’exposition, ont plus de chance de mettre en évidence une relation de cause à effet que des observations isolées tirées d’études uniques. Si le risque de la maladie en question croît avec le degré d’exposition, on considère qu’il y a une forte indication de causalité, bien que l’absence d’une réponse proportionnelle ne prouve pas nécessairement l’absence d’une relation de causalité. La démonstration d’une diminution du risque après l’arrêt ou la diminution de l’exposition des individus ou des populations entières est également en faveur d’une interprétation de causalité des résultats.
Lorsque plusieurs études épidémiologiques fournissent peu ou pas d’indications d’une association entre une exposition et un cancer, on peut en déduire que, dans l’ensemble, elles donnent une indication d’absence de cancérogénicité. Il faut que la possibilité que le biais, la confusion ou la mauvaise classification de l’exposition ou de son effet puisse expliquer les résultats observés soit examinée et exclue avec une certitude raisonnable. Il est important de noter qu’une indication d’absence de pouvoir cancérogène obtenue de cette façon à partir de plusieurs études épidémiologiques ne peut s’appliquer qu’au(x) type(s) de cancer étudié(s) et aux niveaux de doses et aux intervalles entre la première exposition et l’observation de la maladie semblables ou inférieurs à ceux observés dans toutes les études. L’expérience avec le cancer humain montre que, dans certains cas, la période allant de la première exposition au développement d’un cancer clinique est rarement inférieure à vingt ans; les périodes de latence de beaucoup inférieures à trente ans ne peuvent pas constituer une indication d’une absence de cancérogénicité.
Les données relatives à la cancérogénicité dérivées d’études chez l’humain sont classées selon les catégories suivantes:
Indications de cancérogénicité suffisantes: une relation de cause à effet a été établie entre l’exposition à l’agent, au mélange ou aux circonstances d’exposition examinés et le cancer chez l’humain. Une relation positive a été mise en évidence entre l’exposition et la survenue de cancers dans le cadre d’études où les effets du hasard, de biais ou de facteurs de confusion ont pu être exclus avec suffisamment de certitude.
Indications de cancérogénicité limitées: une association positive a été établie entre l’exposition à l’agent, au mélange ou aux circonstances d’exposition considérés et la survenue de cancers. Une interprétation causale de cette association est crédible, mais il n’a pas été possible d’exclure avec suffisamment de certitude que le hasard, un biais ou un facteur de confusion aient pu jouer un rôle.
Indications de cancérogénicité insuffisantes: les études réalisées ne sont pas d’une qualité, d’une concordance ou d’une puissance statistique suffisantes pour permettre de conclure à l’existence ou non d’une relation de cause à effet, ou bien on ne dispose d’aucune donnée sur le cancer chez l’humain.
Indications d’une absence de cancérogénicité: on dispose de plusieurs études suffisantes, couvrant la totalité des niveaux d’exposition connus pour être rencontrés chez l’humain et dont les résultats, concordants, ne font pas ressortir d’association positive entre l’exposition à l’agent, au mélange ou aux circonstances d’exposition considérés et le cancer étudié — et ce, quel que soit le niveau d’exposition examiné. Une conclusion de ce type ne peut s’appliquer qu’aux localisations tumorales, aux conditions et niveaux d’exposition et à la durée d’observation pris en considération dans les études dont on dispose.
La faisabilité de l’évaluation du risque cancérogène présenté par un mélange, un processus, une activité professionnelle ou industrielle à partir d’études épidémiologiques dépend du moment et du lieu. L’exposition, le processus ou l’activité spécifiques considérés comme probablement responsables d’un risque excessif doivent être recherchés et l’évaluation doit en être effectuée de la manière la plus précise possible. La longue période de latence des cancers humains complique l’interprétation des études épidémiologiques. Autre difficulté: le fait que l’humain soit exposé simultanément à plusieurs produits chimiques qui, par leurs interactions, peuvent soit augmenter le risque de néoplasie, soit le diminuer.
Il y a une cinquantaine d’années environ que l’on a commencé à faire des études consistant à exposer des animaux de laboratoire (généralement souris et rats) à des cancérogènes potentiels pour donner une orientation scientifique à l’étude de la cancérogenèse chimique et pallier les inconvénients de l’utilisation exclusive des données épidémiologiques humaines. Dans les monographies du CIRC, les données relatives à la cancérogénicité pour l’animal de laboratoire sont classées selon les catégories suivantes:
Indications de cancérogénicité suffisantes: une relation de cause à effet a été établie entre l’agent ou le mélange examiné et une incidence accrue de néoplasmes malins ou d’une combinaison appropriée de néoplasmes bénins et malins: a) chez deux espèces animales ou plus; ou b) dans le cadre de deux études distinctes ou plus, portant sur une même espèce, effectuées à des moments différents, ou dans des laboratoires différents, ou selon des protocoles différents. Exceptionnellement, une seule étude portant sur une seule espèce peut être considérée comme apportant des indications suffisantes de cancérogénicité lorsqu’une proportion inhabituelle de néoplasmes malins a été observée — tant du point de vue de leur incidence que de leur localisation, du type de tumeur ou de l’âge auquel ils apparaissent.
Indications de cancérogénicité limitées: les données dont on dispose laissent penser qu’il existe un effet cancérogène, mais elles sont limitées et ne permettent pas une évaluation définitive parce que: a) les indications de cancérogénicité se limitent à une seule expérience; ou b) des questions restent en suspens en ce qui concerne la justesse du protocole, de la conduite ou de l’interprétation de l’étude; ou c) l’agent ou le mélange considéré fait augmenter seulement l’incidence des néoplasmes bénins ou des lésions dont le potentiel néoplasique est incertain, ou encore des tumeurs dont la fréquence peut être naturellement élevée chez certaines souches.
Indications de cancérogénicité insuffisantes: les études ne peuvent être interprétées comme prouvant la présence ou l’absence d’un effet cancérogène, parce qu’elles présentent d’importantes faiblesses d’ordre qualitatif ou quantitatif, ou qu’on ne dispose pas de données sur le cancer chez l’animal de laboratoire.
Indications d’une absence de cancérogénicité: on dispose d’un nombre suffisant d’études, portant sur deux espèces au moins, qui montrent que, dans les limites des expériences réalisées, l’agent ou le mélange n’est pas cancérogène. Lorsque les renseignements obtenus suggèrent une absence de cancérogénicité, cette conclusion ne peut s’appliquer qu’aux espèces, aux localisations tumorales et aux niveaux d’exposition étudiés.
Les données sur les effets biologiques chez l’humain qui sont considérées comme utiles sont résumées. Il peut s’agir de données d’ordre toxicologique, pharmacocinétique et métabolique et d’indices de liaisons avec l’ADN, de la persistance de lésions de l’ADN ou d’altérations génétiques chez les sujets exposés. Les données toxicologiques (comme celles sur la cytotoxicité et la régénération, les liaisons aux récepteurs et les effets hormonaux et immunologiques) et celles qui concernent la pharmacocinétique et le métabolisme chez l’animal de laboratoire sont résumées lorsqu’on considère qu’elles jouent un rôle dans l’éventuel mécanisme de l’action cancérogène de l’agent. Les résultats des tests concernant les effets génétiques et apparentés sont récapitulés pour les mammifères, dont l’humain, les cellules de mammifères en culture et les systèmes cellulaires non mammaliens. Les relations structure-activité sont mentionnées lorsqu’elles présentent un intérêt particulier.
Pour l’évaluation de l’agent, du mélange ou de la circonstance d’exposition, toutes les données disponibles portant sur le mécanisme de la cancérogenèse, provenant d’études chez l’humain ou l’animal de laboratoire ou de tests tissulaires et cellulaires, sont récapitulées dans une ou plusieurs des catégories ci-après:
Ces indications ne s’excluent pas les unes les autres, et un agent peut appartenir à plusieurs d’entre elles. C’est ainsi que l’on pourrait répertorier l’action d’un agent sur l’expression de gènes spécifiques à la fois dans la première et dans la seconde catégorie, même si l’on sait de façon assez certaine que ces effets résultent d’une toxicité génétique.
Enfin, tous les éléments d’appréciation sont examinés dans leur ensemble, afin d’arriver à une évaluation globale de la cancérogénicité pour l’humain de l’agent, du mélange ou des circonstances d’exposition considérés. En outre, lorsque des données complémentaires incitent à penser que d’autres produits apparentés, pour lesquels on ne dispose pas d’indications directes de leur capacité d’induire des cancers chez l’humain ou chez l’animal de laboratoire, sont peut-être aussi cancérogènes, on ajoute au compte rendu de l’évaluation un exposé des raisons sur lesquelles se fonde cette conclusion.
L’agent, le mélange ou les circonstances d’exposition sont décrits au moyen des termes désignant l’une des trois catégories ci-après, et l’appartenance à l’un des groupes est établie. Le classement d’un agent, d’un mélange ou de circonstances d’exposition est affaire de jugement scientifique et s’appuie sur le caractère plus ou moins probant des éléments d’appréciation tirés d’études sur l’humain ou l’animal de laboratoire et d’autres informations pertinentes.
L’agent (ou le mélange) est cancérogène pour l’humain. Les circonstances d’exposition donnent lieu à des expositions qui sont cancérogènes pour l’humain.
Cette catégorie n’est utilisée que lorsqu’on dispose d’indications suffisantes de cancérogénicité chez l’être humain. Exceptionnellement, un agent (mélange) peut être placé dans cette catégorie lorsque les indications de cancérogénicité pour l’humain ne sont pas tout à fait suffisantes, mais qu’il existe des indications suffisantes de sa cancérogénicité chez l’animal de laboratoire et de fortes présomptions que l’agent (mélange) agit suivant un mécanisme de cancérogénicité reconnu.
Cette catégorie comporte les agents, mélanges et circonstances d’exposition pour lesquels, au maximum, on a obtenu des indications de cancérogénicité chez l’humain presque suffisantes et, au minimum, on ne dispose d’aucune donnée concernant l’humain, mais on détient des indications suffisantes de cancérogénicité pour l’animal de laboratoire. Lesdits agents, mélanges et circonstances d’exposition sont classés soit dans le groupe 2A (probablement cancérogènes pour l’humain), soit dans le groupe 2B (peut-être cancérogènes pour l’humain) sur la base d’indications épidémiologiques et expérimentales de cancérogénicité et autres renseignements pertinents.
Groupe 2A. L’agent (ou le mélange) est probablement cancérogène pour l’humain. Les circonstances d’exposition donnent lieu à des expositions qui sont probablement cancérogènes pour l’humain. On fait appel à cette catégorie lorsqu’on dispose d’indications limitées de cancérogénicité chez l’être humain et d’indications suffisantes de cancérogénicité chez l’animal de laboratoire. Dans certains cas, un agent (mélange) peut être classé dans cette catégorie lorsqu’on dispose d’indications insuffisantes de cancérogénicité pour l’humain et d’indications suffisantes de cancérogénicité pour l’animal de laboratoire, et de fortes présomptions que la cancérogenèse s’effectue par un mécanisme qui fonctionne également chez l’humain. Exceptionnellement, un agent, un mélange ou une circonstance d’exposition peuvent être classés dans cette catégorie si l’on ne dispose que d’indications limitées de cancérogénicité pour l’humain.
Groupe 2B. L’agent (ou le mélange) est peut-être cancérogène pour l’humain. Les circonstances d’exposition à cet agent donnent lieu à des expositions qui sont peut-être cancérogènes pour l’humain. Cette catégorie est employée lorsqu’on dispose d’indications limitées de cancérogénicité chez l’humain et d’indications insuffisantes de cancérogénicité chez l’animal de laboratoire. On peut aussi y faire appel lorsqu’on dispose d’indications insuffisantes de cancérogénicité pour l’humain, mais d’indications suffisantes pour l’animal de laboratoire. Dans certains cas, on peut classer dans ce groupe un agent, un mélange ou des circonstances d’exposition pour lesquels on a des indications insuffisantes d’une action cancérogène chez l’humain, mais pour lesquels on dispose d’indications limitées de cancérogénicité chez l’animal de laboratoire, corroborées par d’autres données pertinentes.
L’agent (ou le mélange ou les circonstances d’exposition) ne peut pas être classé quant à sa cancérogénicité pour l’humain. Cette catégorie comprend essentiellement les agents, les mélanges et les circonstances d’exposition pour lesquels les indications de cancérogénicité sont insuffisantes chez l’humain et insuffisantes ou limitées chez l’animal de laboratoire.
Exceptionnellement, les agents (ou mélanges) pour lesquels les indications de cancérogénicité sont insuffisantes chez l’humain, mais suffisantes chez l’animal de laboratoire, peuvent être classés dans cette catégorie lorsqu’il existe de fortes présomptions que le mécanisme de cancérogénicité chez l’animal de laboratoire ne fonctionne pas chez l’humain.
L’agent (ou le mélange) n’est probablement pas cancérogène pour l’humain. Relèvent de cette catégorie les agents et mélanges pour lesquels on dispose d’indications suggérant une absence de cancérogénicité chez l’humain ainsi que chez l’animal de laboratoire. Dans certains cas, peuvent être classés dans ce groupe des agents ou des mélanges pour lesquels les indications de cancérogénicité pour l’humain sont insuffisants, mais pour lesquels on dispose d’indications suggérant une absence de cancérogénicité chez l’animal de laboratoire, invariablement et fortement corroborées par une large gamme d’autres données pertinentes.
Les systèmes de classement existants sont encore imparfaits et ne permettent pas de rendre compte de la complexité de la biologie. Ils n’en demeurent pas moins des principes directeurs utiles qui peuvent être modifiés au fur et à mesure que nos connaissances en cancérogenèse progressent. Dans ce classement par catégorie, il est essentiel de faire confiance aux jugements scientifiques établis par les groupes d’experts.
A ce jour on l’a vu, 73 monographies du CIRC ont été publiées ou sont sous presse et 833 agents ou circonstances d’exposition ont été étudiés quant à leur pouvoir cancérogène. Soixante-quinze agents ou expositions ont été classés comme cancérogènes pour l’humain (groupe 1), 59 comme probablement cancérogènes pour l’humain (groupe 2A), 227 comme étant peut-être cancérogènes pour l’humain (groupe 2B) et un comme n’étant probablement pas cancérogène pour l’humain (groupe 4). Pour 471 produits ou expositions, les données épidémiologiques et expérimentales disponibles ne permettent pas d’évaluer leur pouvoir cancérogène chez l’humain (groupe 3).
La modification du préambule, éditée pour la première fois dans le volume 54 des monographies susmentionnées, donne la possibilité de placer dans le groupe 1 un produit dont le pouvoir cancérogène n’est pas suffisamment prouvé du point de vue épidémiologique, alors qu’on dispose d’indications suffisantes de sa cancérogénicité pour l’animal de laboratoire et qu’on le soupçonne fortement d’agir chez l’humain selon un mécanisme en rapport avec la cancérogenèse. Inversement, un agent pour lequel on dispose d’indications insuffisantes de sa cancérogénicité chez l’humain et d’indications suffisantes de sa cancérogénicité chez l’animal de laboratoire, associées à de fortes présomptions que le mécanisme de cancérogenèse ne joue pas chez l’humain, peut être placé dans le groupe 3 au lieu du groupe 2B — peut-être cancérogène pour l’humain — dans lequel il aurait dû normalement être rangé.
L’utilisation des informations concernant le mécanisme d’action a fait l’objet de discussions à l’occasion de trois cas récents: alors qu’on admet généralement que le rayonnement solaire est cancérogène pour l’humain (groupe 1), les études épidémiologiques sur le rôle des rayonnements UVA et UVB des lampes solaires n’apportent qu’une preuve limitée de ce pouvoir cancérogène chez l’humain. Des substitutions spéciales de paires de bases (GCÒTT) ont été observées au niveau des gènes suppresseurs de tumeur p53 dans les tumeurs épithéliales à des sites exposés au soleil chez l’humain. Bien que les rayonnements UV puissent induire des transitions similaires dans certains systèmes expérimentaux et que les UVB, UVA et UVC soient cancérogènes chez l’animal de laboratoire, les données disponibles sur leur mécanisme d’action n’ont pas été considérées comme étant suffisamment probantes pour permettre au groupe de travail de classer les UVB, UVA et UVC dans un groupe supérieur au groupe 2A (CIRC, 1992). Dans une étude publiée après la réunion du groupe de travail (Kress et coll., 1992), des transitions CCÒTT au niveau du gène p53 ont été mises en évidence dans les tumeurs cutanées induites par les UVB chez la souris, ce qui donne à penser que les UVB devraient également être classés comme cancérogènes pour l’humain (groupe 1).
Le second cas où on a envisagé de placer un agent dans le groupe 1 en l’absence de preuve épidémiologique suffisante est celui du 4,4’-méthylène-bis(2-chloroaniline) (MOCA). Le MOCA est cancérogène chez le chien et les rongeurs et a un pouvoir génotoxique significatif. Il se lie à l’ADN par réaction avec le N-hydroxy MOCA et les adduits formés dans les tissus cibles chez l’animal ont été retrouvés également au niveau des cellules urothéliales d’un petit nombre de personnes exposées. Après de longues discussions sur la possibilité d’un reclassement, le groupe de travail a finalement décidé de ranger le produit dans le groupe 2A, probablement cancérogène pour l’humain (CIRC, 1993).
Troisième et dernier exemple, celui de l’oxyde d’éthylène (CIRC, 1994b). Les études épidémiologiques dont on disposait fournissaient des indications limitées de sa cancérogénicité pour l’humain, les études chez l’animal ayant apporté des indications suffisantes de son pouvoir cancérogène. Au vu des autres données pertinentes, l’oxyde d’éthylène a été classé comme cancérogène chez l’humain (groupe 1) pour les raisons suivantes: 1) il induit une augmentation sensible, persistante et dose-dépendante de la fréquence des aberrations chromosomiques et des échanges de chromatides sœurs dans les lymphocytes périphériques, et de micronoyaux dans les cellules de la moelle osseuse des travailleurs exposés; 2) il est associé à des tumeurs malignes du système lymphatique et hématopoïétique chez l’humain et chez l’animal de laboratoire; 3) il induit une augmentation dose-dépendante de la fréquence des adduits à l’hémoglobine chez les personnes exposées et des augmentations, dose-dépendantes elles aussi, du nombre d’adduits à l’ADN et à l’hémoglobine chez les rongeurs exposés; 4) il induit des mutations géniques et des translocations héréditaires dans les cellules germinales de rongeurs exposés; et 5) c’est un mutagène et un clastogène puissant à tous les niveaux phylogéniques. Pour toutes ces raisons, l’oxyde d’éthylène a donc été classé dans la catégorie des agents cancérogènes pour l’homme (groupe 1).
Le préambule précité donne la possibilité de classer un agent dans le groupe 3 (au lieu du groupe 2B où il devrait être normalement rangé). Aucun groupe de travail ne s’est prévalu de cette possibilité lorsqu’il existait à la fois des indications suffisantes de cancérogénicité d’un agent pour l’animal de laboratoire et une forte présomption que le mécanisme d’action responsable chez l’animal ne pouvait se produire chez l’humain. Une telle possibilité aurait pu être envisagée pour le d-limonène si on avait eu des indications suffisantes de sa cancérogénicité chez l’animal, puisqu’on dispose de données donnant à penser que les tumeurs rénales observées chez le rat mâle apparaissent liées à la production d’α2-microglobuline non synthétisée.
Parmi les nombreux produits chimiques classés comme prioritaires par un groupe de travail spécial en décembre 1993, on a mis en évidence des mécanismes d’action communs supposés intrinsèques ou identifié certaines classes de produits sur la base de leurs propriétés biologiques. Le groupe de travail a recommandé, qu’avant de procéder à l’évaluation de produits tels que les proliférateurs de peroxysomes, les fibres, les poussières et les agents thyrostatiques dans le programme des monographies, il convenait de créer des groupes spéciaux qui seraient chargés de dresser un bilan des connaissances sur les mécanismes d’action particuliers de ces agents.
Aflatoxines, mélanges naturels [1402-68-2] (1993)
Amiante [1332-21-4] (1987)
4-Aminobiphényle [92-67-1] (1987)
Arsenic [7440-38-2] et ses composés (1987)2
Azathioprine [446-86-6] (1987)
Benzène [71-43-2] (1987)
Benzidine [92-87-5] (1987)
Béryllium [7440-41-7] et ses composés (1993)3
Bis(2-chloroéthyl)-2-naphthylamine (Chlornaphazine) [494-03-1] (1987)
Bis(chlorométhyl)éther [542-88-1] et chlorométhyl- méthyléther [107-30-2] (qualité technique) (1987)
Cadmium [7440-43-9] et ses composés (1993)3
Chlorambucile [305-03-3] (1987)
1-(2-Chloroéthyl)-3-(4-méthylcyclohexyl)-1-nitrosourée (Méthyl-CCNU; Sémustine) [13909-09-6] (1987)
Chlorure de vinyle [75-01-4] (1987)
Ciclosporine [79217-60-0] (1990)
Composés du chrome [VI] (1990)3
Composés du nickel (1990) 3
Contraceptifs oraux en association (1999)4
Contraceptifs oraux séquentiels (1987)
Cyclophosphamide [50-18-0] [6055-19-2] (1987)
Diéthylstilbœstrol [56-53-1] (1987)
Diméthanesulfonate de 1,4-butanediol (Busulphan; Myleran) [55-98-1] (1987)
Erionite [66733-21-9] (1987)
Gaz moutarde (Moutarde sulfurée) [505-60-2] (1987)
Helicobacter pylori (infection) (1994)
Melphalane [148-82-3] (1987)
8-Méthoxypsoralène (Méthoxsalène) [298-81-7] associé au rayonnement ultraviolet A (1987)
MOPP (traitement associé utilisant moutarde azotée, vincristine, procarbazine et prednisone) et autres chimio- thérapies associées utilisant des agents alkylants (1987)
2-Naphtylamine [91-59-8] (1987)
Œstrogénothérapie de substitution (1999)
Œstrogènes non stéroïdiens (1987)2
Œstrogènes stéroïdiens (1987)2
Opistorchis viverrini (infestation) (1994)
Oxyde d’éthylène [75-21-8] (1994)6
Radon [10043-92-2] et ses produits de filiation (1988)
Rayonnement solaire (1992)
Schistosoma hæmatobium (infestation) (1994)
Silice cristalline [14808-60-7] (inhalée sous forme de quartz ou de cristobalite de source professionnelle) (1997)
Talc contenant des fibres asbestiformes (1987)
Tamoxifène [10540-29-1] (1996)5
Tétrachloro-2,3,7,8 dibenzo-p-dioxine [1746-01-6] (1997)6
Thiotépa [52-24-4] (1990)
Tréosulfan [299-75-2] (1987)
Virus d’Epstein-Barr (1997)
Virus de l’hépatite B (VHB) (infection chronique) (1994)
Virus de l’hépatite C (VHC) (infection chronique) (1994)
Virus de l’immunodéficience humaine de type 1 (VIH 1) (infection) (1996)
Virus du papillome humain de type 16 (VPH 16) (1995)
Virus du papillome humain de type 18 (VPH 18) (1995)
Virus humain de la leucémie à cellules T, type I (HTLV-1) (1996)
Boissons alcooliques (1988)
Brais de houille [65996-93-2] (1987)
Fumée de tabac (1987)
Goudrons de houille [8007-45-2] (1987)
Huiles de schiste [68308-34-9] (1987)
Huiles minérales, peu ou non raffinées (1987)
Mastication de bétel avec tabac (1987)
Mélanges analgésiques contenant de la phénacétine (1987)
Poisson salé (façon chinoise) (1993)
Poussières de bois (1995)
Produits du tabac non fumé (1987)
Suies (1987)
Aluminium (production) (1987)
Auramine (fabrication) (1987)
Brouillards d’acides inorganiques forts contenant de l’acide sulfurique (exposition professionnelle) (1992)
Caoutchouc (industrie) (1987)
Charbon (gaséification) (1987)
Chaussures (fabrication et réparation) (1987)
Coke (production) (1987)
Ebénisterie et menuiserie (1987)
Hématite (extraction souterraine avec exposition concomitante au radon) (1987)
Isopropanol (fabrication) (procédé aux acides forts) (1987)
Magenta (fabrication) (1993)
Métallurgie du fer et de l’acier (1987)
Peintres (exposition professionnelle) (1989)
Acrylamide [79-06-1] (1994)7
Adriamycine [23214-92-8] (1987)7
Azacitidine [320-67-2] (1990)7
Benzo[a]anthracène [56-55-3] (1987)7
Benzo[a]pyrène [50-32-8] (1987)7
Bischloroéthyl-nitrosourée (BCNU) [154-93-8] (1987)
Bromure de vinyle [593-60-2] (1999)7
1,3-Butadiène [106-99-0] (1999)
Captafol [2425-06-1] (1991)7
Chloramphénicol [56-75-7] (1990)7
1-(2-Chloroéthyl)-3-cyclohexyl-1-nitrosourée (CCNU) [13010-47-4] (1987)7
p-Chloro-o-toluidine [95-69-2] et ses sels d’acides forts (1990)3
Chlorozotocine [54749-90-5] (1990)7
Chlorure de diméthylcarbamoyle [79-44-7] (1999)7
Cisplatine [15663-27-1] (1987)7
Clonorchis sinensis (infestation) (1994)7
Colorants à base de benzidine (1987)7
Dibenzo[a,h]anthracène [53-70-3] (1987)7
1,2-Dibromoéthane [106-93-4] (1999)7
1,2-Diméthylhydrazine [540-73-8] (1999)
Epichlorohydrine [106-89-8] (1999)7
N-Ethyl-N-nitrosourée [759-73-9] (1987)7
Fluorure de vinyle [75-02-5] (1995)
Formaldéhyde [50-00-0] (1995)
Herpès virus du sarcome de Kaposi/herpès virus humain no 8 (1997)
Hydrochlorure de procarbazine [366-70-1] (1987)7
IQ (2-Amino-3-méthylimidazo[4,5-f]quinoléine) [76180-96-6] (1993)7
Méthanesulfonate de méthyle [66-27-3] (1999)7
5-Méthoxypsoralène [484-20-8] (1987)7
4,4´-Méthylène bis(2-chloroaniline) (MOCA) [101-14-4] (1993)7
N-Méthyl-N´-nitro-N-nitrosoguanidine (MNNG) [70-25-7] (1987)7
N-Méthyl-N-nitrosourée [684-93-5] (1987)7
Moutarde azotée [51-75-2] (1987)
N-Nitrosodiéthylamine [55-18-5] (1987)7
N-Nitrosodiméthylamine [62-75-9] (1987)7
7,8-Oxyde de styrène [96-09-3] (1994)7
Phénacétine [62-44-2] (1987)
Phosphate de tris(2,3 dibromopropyle) [126-72-7] (1999)7
Rayonnements ultraviolets A (1992)7
Rayonnements ultraviolets B (1992)7
Rayonnements ultraviolets C (1992)7
Stéroïdes androgéniques (anabolisants) (1987)
Sulfate de diéthyle [64-67-5] (1999)
Sulfate de diméthyle [77-78-1] (1999)7
Tétrachloroéthylène [127-18-4] (1995)
Toluènes α-chlorés (benzotrichlorure [98-07-7], chlorure de benzal [98-87-3], chlorure de benzyle [100-44-7]) et chlorure de benzoyle [98-88-4] (expositions mixtes) (1999)
Trichloroéthylène [79-01-6] (1995)
1,2,3-Trichloropropane [96-18-4] (1995)
Virus du papillome humain de type 31 (1995)
Virus du papillome humain de type 33 (1995)
Biphényles polychlorés [1336-36-3] (1987)
Créosotes [8001-58-9] (1987)
Gaz d’échappement de moteur diesel (1989)
Insecticides non arsenicaux (expositions professionnelles lors de la pulvérisation et de l’application) (1991)
Maté chaud (1991)
Coiffeurs (exposition professionnelle) (1993)
Lampes et tables à bronzer (utilisation) (1992)
Raffinage du pétrole (exposition professionnelle) (1989)
Verrerie d’art, verre creux et verre moulé (fabrication) (1993)
A-α-C (2-Amino-9H-pyrido[2,3-b]indole) [26148-68-5] (1987)
Acétaldéhyde [75-07-0] (1999)
Acétamide [60-35-5] (1999)
Acétate de médroxyprogestérone [71-58-9] (1987)
Acétate de méthylazoxyméthanol [592-62-1] (1987)
Acétate de vinyle [108-05-4] (1995)
Acide caféique [331-39-5] (1993)
Acide chlorendique [115-28-6] (1990)
Acide nitrilotriacétique [139-13-9] et ses sels (1999)3
Acrylate d’éthyle [140-88-5] (1999)
Acrylonitrile [107-13-1] (1999)
AF-2 [2-(2-Furyl)-3 (5-nitro-2-furyl)acrylamide] [3688-53-7] (1987)
Aflatoxine M1 [6795-23-9] (1993)
p-Aminoazobenzène [60-09-3] (1987)
o-Aminoazotoluène [97-56-3] (1987)
2-Amino-5-(5-nitro-2- furyl)-1,3,4-thiadiazole [712-68-5] (1987)
Amitrole [61-82-5] (1987)
o-Anisidine [90-04-0] (1999)
Aramite® [140-57-8] (1987)
Auramine [492-80-8] (qualité technique) (1987)
Azasérine [115-02-6] (1987)
Aziridine [151-56-4] (1999)
Benzo[b]fluoranthène [205-99-2] (1987)
Benzo[j]fluoranthène [205-82-3] (1987)
Benzo[k]fluoranthène [207-08-9] (1987)
Benzofuranne [271-89-6] (1995)
Bléomycines [11056-06-7] (1987)8
Bleu direct CI-15 [2429-74-5] (1993)
Bleu dispersé 1 [2475-45-8] (1990)
Bleu HC 1 [2784-94-3] (1993)
Bleu Trypan [72-57-1] (1987)
Bromate de potassium [7758-01-2] (1999)
Bromodichlorométhane [75-27-4] (1999)
Butyl hydroxyanisole (BHA) [25013-16-5] (1987)
β-Butyrolactone [3068-88-0] (1999)
Catéchol [120-80-9] (1999)
Chlordane [57-74-9] (1991)
Chlordécone (Képone) [143-50-0] (1987)
Chlorhydrate de phénazopyridine [136-40-3] (1987)
Chlorhydrate de phénoxybenzamine [63-92-3] (1987)
p-Chloroaniline [106-47-8] (1993)
Chloroforme [67-66-3] (1999)
1-Chloro-2-méthylpropène [513-37-1] (1995)
4-Chloro-o-phénylènediamine [95-83-0] (1987)
Chloroprène [126-99-8] (1999)
Cobalt [7440-48-4] et ses composés (1991)3
Complexe fer-dextran [9004-66-4] (1987)
Composés de méthylmercure (1993)3
Contraceptifs, uniquement progestatifs (1999)
p-Crésidine [120-71-8] (1987)
Cycasine [14901-08-7] (1987)
Dacarbazine [4342-03-4] (1987)
Dantrone (Chrysazine; 1,8-Dihydroxyanthraquinone) [117-10-2] (1990)
Daunomycine [20830-81-3] (1987)
DDT [p,p´-DDT, 50-29-3] (1991)
N,N´-Diacétylbenzidine [613-35-4] (1987)
2-4-Diaminoanisole [615-05-4] (1987)
4,4´-Diaminodiphényléther [101-80-4] (1987)
2,4-Diaminotoluène [95-80-7] (1987)
Dibenzo[a,h]acridine [226-36-8] (1987)
Dibenzo[a,j]acridine [224-42-0] (1987)
7H-Dibenzo[c,g]carbazole [194-59-2] (1987)
Dibenzo[a,e]pyrène [192-65-4] (1987)
Dibenzo[a,h]pyrène [189-64-0] (1987)
Dibenzo[a,i]pyrène [189-55-9] (1987)
Dibenzo[a,l]pyrène [191-30-0] (1987)
1,2-Dibromo-3-chloropropane [96-12-8] (1999)
p-Dichlorobenzène [106-46-7] (1999)10
3,3´-Dichlorobenzidine [91-94-1] (1987)
3,3´-Dichloro-4,4´-diaminodiphényléther [28434-86-8] (1987)
1,2-Dichloroéthane [107-06-2] (1999)
Dichlorométhane (Chlorure de méthylène) [75-09-2] (1999)
1,3-Dichloropropène [542-75-6] (qualité technique) (1999)
Dichlorvos [62-73-7] (1991)
1,2-Diéthylhydrazine [1615-80-1] (1999)
Diglycidylrésorcinoléther [101-90-6] (1999)
Dihydrosafrol [94-58-6] (1987)
Diisocyanates de toluène [26471-62-5] (1999)
3,3´-Diméthoxybenzidine (o-Dianisidine) [119-90-4] (1987)
p-Diméthylaminoazobenzène [60-11-7] (1987)
trans-2[(Diméthylamino)méthylimino]5-[2-(5-nitro-2-furyl)- vinyle]-1,3,4-oxadiazole [25962-77-0] (1987)
2,6-Diméthylaniline (2,6-Xylidine) [87-62-7] (1993)
3,3´-Diméthylbenzidine (o-Toluidine) [119-93-7] (1987)
1,1-Diméthylhydrazine [57-14-7] (1999)
3,7-Dinitrofluoranthène [105735-71-5] (1996)
3,9-Dinitrofluoranthène [22506-53-2] (1996)
1,6-Dinitropyrène [42397-64-8] (1989)
1,8-Dinitropyrène [42397-65-9] (1989)
2,4-Dinitrotoluène [121-14-2] (1996)
2,6-Dinitrotoluène [606-20-2] (1996)
1,4-Dioxane [123-91-1] (1999)
1,2-Epoxybutane [106-88-7] (1999)8
Ethylènethiourée [96-45-7] (1987)
Fibres céramiques (1988)
2-(2-Formylhydrazino)-4-(5-nitro-2-furyl)thiazole [3570-75-0] (1987)
Fougère arborescente (1987)
Furanne [110-00-9] (1995)
Glu-P-1 (2-Amino-6-méthyldipyrido[1,2-a:3´,2´-d]imidazole) [67730-11-4] (1987)
Glu-P-2 (2-Aminodipyrido[1,2-a:3´,2´-d]imidazole) [67730-10-3] (1987)
Glycidaldéhyde [765-34-4] (1999)
Griséofulvine [126-07-8] (1987)
Heptachlore [76-44-8] (1991)
Herbicides chlorophénoxy (1987)
Hexachlorobenzène [118-74-1] (1987)
Hexachlorocyclohexanes (1987)
Hexachloroéthane [67-72-1] (1999)
Hexaméthylphosphoramide [680-31-9] (1999)
Hydrazine [302-01-2] (1999)
Indeno[1,2,3-cd]pyrène [193-39-5] (1987)
Isoprène [78-79-5] (1999)
Laine de laitier (1988)
Laine de roche (1988)
Laine de verre (1988)
Lasiocarpine [303-34-4] (1987)
Magenta [632-99-5] (contenant du Rouge basique CI-9) (1993)
MeA-α-C (2-Amino-3-méthyl-9H-pyrido[2,3-b]indole) [68006-83-7] (1987)
MeIQ (2-Amino-3,4-diméthylimidazo[4,5-f]quinoléine) [77094-11-2] (1993)
MeIQx (2-Amino-3,8-diméthylimidazo[4,5-f]quinoxaline) [77500-04-0] (1993)
Merphalane [531-76-0] (1987)
Méthanesulfonate d’éthyle [62-50-0] (1987)
2-Méthylaziridine (Propylèneimine) [75-55-8] (1999)
5-Méthylchrysène [3697-24-3] (1987)
4,4´-Méthylène-bis(2-méthylaniline) [838-88-0] (1987)
4,4´-Méthylènedianiline [101-77-9] (1987)
2-Méthyl-1-nitroanthraquinone [129-15-7] (pureté non connue) (1987)
N-Méthyl-N-nitrosouréthane [615-53-2] (1987)
Méthylthiouracile [56-04-2] (1987)
Métronidazole [443-48-1] (1987)
Mirex [2385-85-5] (1987)
Mitomycine C [50-07-7] (1987)
Monocrotaline [315-22-0] (1987)
5-(Morpholinométhyl)-3-[(5-nitrofurfurylidène)amino]-2- oxazolidinone [3795-88-8] (1987)
Moutarde d’uracile [66-75-1] (1987)
Nafénopine [3771-19-5] (1987)
Nickel (métal) [7440-02-0] (1990)
Niridazole [61-57-4] (1987)
5-Nitroacénaphthène [602-87-9] (1987)
2-Nitroanisole [91-23-6] (1996)
Nitrobenzène [98-95-3] (1996)
6-Nitrochrysène [7496-02-8] (1989)
Nitrofène [1836-75-5] (qualité technique) (1987)
2-Nitrofluorène [607-57-8] (1989)
1-[(5-Nitrofurfurylidène)amino]-2-imidazolidinone [555-84-0] (1987)
N-[4-(5-Nitro-2-furyl)-2-thiazolyl]acétamide [531-82-8] (1987)
2-Nitropropane [79-46-9] (1999)
1-Nitropyrène [5522-43-0] (1989)
4-Nitropyrène [57835-92-4] (1989)
N-Nitrosodi-n-butylamine [924-16-3] (1987)
N-Nitrosodiéthanolamine [1116-54-7] (1987)
N-Nitrosodi-n-propylamine [621-64-7] (1987)
3-(N-Nitrosométhylamino)propionitrile [60153-49-3] (1987)
4-(N-Nitrosométhylamino)-1-(3-pyridyl)-1-butanone (NNK) [64091-91-4] (1987)
N-Nitrosométhyléthylamine [10595-95-6] (1987)
N-Nitrosométhylvinylamine [4549-40-0] (1987)
N-Nitrosomorpholine [59-89-2] (1987)
N´-Nitrosonornicotine [16543-55-8] (1987)
N-Nitrosopipéridine [100-75-4] (1987)
N-Nitrosopyrrolidine [930-55-2] (1987)
N-Nitrososarcosine [13256-22-9] (1987)
Noir de carbone [1333-86-4] (1996)
Ochratoxine A [303-47-9] (1993)
Orangé huileux SS [2646-17-5] (1987)
Oxazépam [604-75-1] (1996)
N-Oxyde de moutarde azotée [126-85-2] (1987)
Oxyde de propylène [75-56-9] (1994)
Palygorskite (attapulgite) [12174-11-7] (fibres longues (> 5 µm) (1997)
Panfuran-S [794-93-4] (contenant de la dihydroxyméthyl- furatrizine) (1987)
Phénobarbital [50-06-6] (1987)
Phénylglycidyléther [122-60-1] (1999)
o-Phénylphénate de sodium [132-27-4] (1999)
Phénytoïne [57-41-0] (1996)
PhIP (2-Amino-1-méthyl-6-phénylimidazo[4,5-b]pyridine) [105650-23-5] (1993)
Phtalate de di(2-éthylhexyle) [117-81-7] (1987)
Plomb [7439-92-1] et dérivés inorganiques du plomb (1987)3
Polychlorophénols et leurs sels de sodium (expositions mixtes) (1999)
Ponceau MX [3761-53-3] (1987)
Ponceau 3R [3564-09-8] (1987)
Progestatifs (1987)
1,3-Propanesultone [1120-71-4] (1999)
β-Propiolactone [57-57-8] (1999)
Propylthiouracile [51-52-5] (1987)
Rouge acide CI-114 [6459-94-5] (1993)
Rouge basique CI-9 [569-61-9] (1993)
Rouge citrus 2 [6358-53-8] (1987)
Safrole [94-59-7] (1987)
Schistosoma japonicum (infestation) (1994)
Stérigmatocystine [10048-13-2] (1987)
Streptozotocine [18883-66-4] (1987)
Styrène [100-42-5] (1994)8
Sulfallate [95-06-7] (1987)
Sulfate de diisopropyle [2973-10-6] (1999)
Tétrachloroisophtalonitrile (Chlorothalonil) [1897-45-6] (1999)
Tétrachlorure de carbone [56-23-5] (1999)
Tétrafluoroéthylène [116-14-3] (1999)
Tétranitrométhane [509-14-8] (1996)
Thérapie œstrogéno-progestative de substitution (1999)
Thioacétamide [62-55-5] (1987)
4,4´-Thiodianiline [139-65-1] (1987)
Thiourée [62-56-6] (1987)
o-Toluidine [95-53-4] (1987)
Toxines dérivées du Fusarium moniliforme (1993)
Trichlorométhine (Chlorhydrate de trimustine) [817-09-4] (1990)
Trioxyde d’antimoine [1309-64-4] (1989)
Trp-P-1 (3-Amino-1,4-diméthyl-5H-pyrido[4,3-b]indole) [62450-06-0] (1987)
Trp-P-2 (3-Amino-1-méthyl-5H-pyrido[4,3-b]indole) [62450-07-1] (1987)
Uréthane [51-79-6] (1987)
4-Vinylcyclohexène [100-40-3] (1994)
4-Vinylcyclohexène diépoxyde [106-87-6] (1994)
Violet de benzyle 4B [1694-09-3] (1987)
Virus de l’immunodéficience humaine (VIH) de type 2 (infection) (1996)
Virus du papillome humain (VPH) autres que les types 16, 18, 31 et 33 (1995)
Biphényles polybromés [Firemaster BP-6, 59536-65-1] (1987)
Bitumes [8052-42-4], extraits, raffinés à la vapeur et raffinés à l’air (1987)
Café (vessie urinaire) (1991)9
Carburants diesel marins (1989)8
Carrageenan [9000-07-1] dégradé (1987)
Essence (1989)8
Fuel résiduel (lourd) (1989)
Fumées de soudage (1990)
Gaz d’échappement de moteur, essence (1989)
Légumes au vinaigre (condiment asiatique traditionnel) (1993)
Paraffines chlorées dont la longueur moyenne de la chaîne carbonée est de C12 et le taux moyen de chloration de 60% environ (1990)
Toxaphène (camphènes polychlorés) [8001-35-2] (1987)
Charpenterie et menuiserie (1987)
Industrie textile (fabrication) (exposition professionnelle) (1990)
Nettoyage à sec (exposition professionnelle) (1995)
Procédés d’impression (exposition professionnelle) (1996)
Acétate de benzyle [140-11-4] (1999)
Acide acrylique [79-10-7] (1999)
p-Acide aminobenzoïque (4-Acide aminobenzoïque) [150-13-0] (1987)
Acide 11-aminoundécanoïque [2432-99-7] (1987)
Acide anthranilique [118-92-3] (1987)
Acide chlorhydrique [7647-01-0] (1992)
Acide dichloroacétique [79-43-6] (1995)
Acide cis-9,10-époxystéarique [2443-39-2] (1999)
N-Acide nitrosofolique [29291-35-8] (1987)
Acide parasorbique [10048-32-5] (1987)
Acide pénicillique [90-65-3] (1987)
Acide polyacrylique [9003-01-4] (1987)
Acide shikimique [138-59-0] (1987)
Acide tannique [1401-55-4] et tanins (1987)
Acide trichloroacétique [76-03-9] (1995)
Acroléine [107-02-8] (1995)
Acrylate de n-butyle [141-32-2] (1999)
Acrylate de 2-éthylhexyle [103-11-7] (1994)
Acrylate de méthyle [96-33-3] (1999)
Actinomycine D [50-76-0] (1987)
Agaritine [2757-90-6] (1987)
Aldicarb [116-06-3] (1991)
Aldrine [309-00-2] (1987)
Amarante [915-67-3] (1987)
5-Aminoacénaphtène [4657-93-6] (1987)
2-Aminoanthraquinone [117-79-3] (1987)
1-Amino-2-méthylanthraquinone [82-28-0] (1987)
2-Amino-4-nitrophénol [99-57-0] (1993)
2-Amino-5-nitrophénol [121-88-0] (1993)
4-Amino-2-nitrophénol [119-34-6] (1987)
2-Amino-5-nitrothiazole [121-66-4] (1987)
Ampicilline [69-53-4] (1990)
Anesthésiques volatils (1987)
Angélicine [523-50-2] et exposition aux rayonnements ultraviolets A (1987)
Anhydride succinique [108-30-5] (1987)
Aniline [62-53-3] (1987)
p-Anisidine [104-94-9] (1987)
Anthanthrène [191-26-4] (1987)
Anthracène [120-12-7] (1987)
Anthranilate de cinnamyle [87-29-6] (1987)
Apholate [52-46-0] (1987)
Atrazine [1912-24-9] (1999)11
Aurothioglucose [12192-57-3] (1987)
2-(1-Aziridinyl)éthanol [1072-52-2] (1987)
Aziridylbenzoquinone [800-24-8] (1987)
Azobenzène [103-33-3] (1987)
Benzo[a]acridine [225-11-6] (1987)
Benzo[c]acridine [225-51-4] (1987)
Benzo[ghi]fluoranthène [203-12-3] (1987)
Benzo[a]fluorène [238-84-6] (1987)
Benzo[b]fluorène [243-17-4] (1987)
Benzo[c]fluorène [205-12-9] (1987)
Benzo[ghi]pérylène [191-24-2] (1987)
Benzo[c]phénanthrène [195-19-7] (1987)
Benzo[e]pyrène [192-97-2] (1987)
1,4-Benzoquinone-dioxine [105-11-3] (1999)
Bis(2-chloro-1-méthyléthyl)éther [108-60-1] (1999)
Bis(2-chloroéthyl)éther [111-44-4] (1999)
1,2-Bis-(chlorométhoxy)éthane [13483-18-6] (1999)
1,4-Bis-(chlorométhoxyméthyl)benzène [56894-91-8] (1999)
Bis-(2,3-époxycyclopentyl)éther [2386-90-5] (1999)
Bisulfites (1992)
Bleu brillant FCF (sel disodique) [3844-45-9] (1987)
Bleu Evans [314-13-6] (1987)
Bleu HC 2 [33229-34-4] (1993)
Bleu VRS [129-17-9] (1987)
Bromochloroacétonitrile [83463-62-1] (1999)
Bromoéthane [74-96-4] (1999)
Bromoforme [75-25-2] (1999)
Bromure de méthyle [74-83-9] (1999)
Brun Soudan RR [6416-57-5] (1987)
Butoxyde de pipéronyle [51-03-6] (1987)
γ-Butyrolactone [96-48-0] (1999)
Caféine [58-08-2] (1991)
Cantharidine [56-25-7] (1987)
Captan [133-06-2] (1987)
Carbamate de méthyle [598-55-0] (1987)
Carbaryle [63-25-2] (1987)
Carbazole [86-74-8] (1999)
3-Carbéthoxypsoralène [20073-24-9] (1987)
Carmoisine [3567-69-9] (1987)
Carrageenan naturel [9000-07-1] (1987)
Chloral [75-87-6] (1995)
Chlordiméform [6164-98-3] (1987)
Chlorhydrate de pronétalol [51-02-5] (1987)
Chlorhydrate de semicarbazide [563-41-7] (1987)
Chlorite de sodium [7758-19-2] (1991)
Chloroacétonitrile [107-14-2] (1999)
Chlorodibromométhane [124-48-1] (1999)
Chlorodifluorométhane [75-45-6] (1999)
Chloroéthane [75-00-3] (1999)
Chlorofluorométhane [593-70-4] (1999)
3-Chloro-2-méthylpropylène [563-47-3] (1995)
Chloronitrobenzènes (mélange d’isomères) [88-73-3; 121-73-3; 100-00-5] (1996)
4-Chloro-m-phénylènediamine [5131-60-2] (1987)
Chloropropham [101-21-3] (1987)
Chloroquine [54-05-7] (1987)
2-Chloro-1,1,1-trifluoroéthane [75-88-7] (1999)
Chlorure d’acriflavinium [8018-07-3] (1987)
Chlorure d’allyle [107-05-1] (1999)
Chlorure de méthyle [74-87-3] (1999)
Chlorure de vinylidène [75-35-4] (1999)
Cholestérol [57-88-5] (1987)
Chrome métallique [7440-47-3] (1990)
Chrysène [218-01-9] (1987)
Chrysoïdine [532-82-1] (1987)
Cimétidine [51481-61-9] (1990)
Citrate de clomiphène [50-41-9] (1987)
Citrinine [518-75-2] (1987)
Clofibrate [637-07-0] (1996)
Complexe fer-dextrine [9004-51-7] (1987)
Complexe-fer-sorbitol-acide citrique [1338-16-5] (1987)
Composés du chrome III (1990)
Composés organiques du plomb [75-74-1], [78-00-2] (1987)
Copolymères acrylonitrile-butadiène-styrène (1987)
Copolymères chlorure de vinyle-acétate de vinyle [9003-22-9] (1987)
Copolymères chlorure de vinylidène-chlorure de vinyle [9011-06-7] (1987)
Copolymères styrène-acrylonitrile [9003-54-7] (1987)
Copolymères styrène-1,3-butadiène [9003-55-8] (1987)
Coronène [191-07-1] (1987)
Coumarine [91-64-5] (1987)
m-Crésidine [102-50-1] (1987)
Crotonaldéhyde [4170-30-3] (1995)
Cyclamates [cyclamate de sodium, 139-05-9] (1999)
Cyclochlorotine [12663-46-6] (1987)
Cyclohexanone [108-94-1] (1999)
Cyclopenta[cd]pyrène [27208-37-3] (1987)
Dapsone [80-08-0] (1987)
Deltaméthrine [52918-63-5] (1991)
Diacétylaminoazotoluène [83-63-6] (1987)
Diallate [2303-16-4] (1987)
1,5-Diaminonaphtalène [2243-62-1] (1987)
1,2-Diamino-4-nitrobenzène [99-56-9] (1987)
1,4-Diamino-2-nitrobenzène [5307-14-2] (1993)
2,5-Diaminotoluène [95-70-5] (1987)
Diazépam [439-14-5] (1996)
Diazométhane [334-88-3] (1987)
Dibenzo[a,c]anthracène [215-58-7] (1987)
Dibenzo[a,j]anthracène [224-41-9] (1987)
Dibenzo-p-dioxine (1997)
Dibenzo-p-dioxines polychlorées (autres que 2,3,7,8-tétra-chlorodibenzo-p-dioxine) (1997)
Dibenzo[a,e]fluoranthène [5385-75-1] (1987)
Dibenzofuranes polychlorés (1997)
Dibenzo[h,rst]pentaphène [192-47-2] (1987)
Dibromoacétonitrile [3252-43-5] (1999)
Dichlorhydrate de mannomustine [551-74-6] (1987)
Dichloroacétonitrile [3018-12-0] (1999)
Dichloroacétylène [7572-29-4] (1999)
1,2-Dichlorobenzène [95-50-1] (1999)
1,3-Dichlorobenzène [541-73-1] (1999)
4,4´-Dichlorobenzilate d’éthyle (Chlorobenzilate) [510-15-6] (1987)
trans-1,4-Dichloro-2-butène [110-57-6] (1999)
2,6-Dichloro-p-phénylènediamine [609-20-1] (1987)
1,2-Dichloropropane [78-87-5] (1999)
Dicofol [115-32-2] (1987)
Dieldrine [60-57-1] (1987)
Diéthyldithiocarbamate de sélénium [5456-28-0] (1987)
Diéthyldithiocarbamate de sodium [148-18-5] (1987)
Diéthyldithiocarbamate de tellure [20941-65-5] (1987)
Di(2-éthylhexyle)adipate [103-23-1] (1987)
Diglycidyléther du bisphénol A (Araldite®) [1675-54-3] (1999)
Dihydroxyméthylfuratrizine [794-93-4] (1987)
Diméthoxane [828-00-2] (1987)
3,3´-Diméthoxybenzidine-4-4´-diisocyanate [91-93-0] (1987)
p-Diméthylaminoazobenzènediazosulfonate de sodium [140-56-7] (1987)
4,4´-Diméthylangélicine [22975-76-4] et exposition aux rayonnements ultraviolets A (1987)
4,5´-Diméthylangélicine [4063-41-6] et exposition aux rayonnements ultraviolets A (1987)
N,N-Diméthylaniline [121-69-7] (1993)
Diméthyldithiocarbamate de sélénium [144-34-3] (1987)
Diméthylformamide [68-12-2] (1999)
1,4-Diméthylphénanthrène [22349-59-3] (1987)
1,3-Dinitropyrène [75321-20-9] (1989)
Dinitrosopentaméthylènetétramine [101-25-7] (1987)
3,5-Dinitrotoluène [618-85-9] (1996)
Dioxyde de soufre [7446-09-5] (1992)
Dioxyde de titane [13463-67-7] (1989)
2,4´-Diphényldiamine [492-17-1] (1987)
Disulfiram [97-77-8] (1987)
Dithranol [1143-38-0] (1987)
Doxéfazépam [40762-15-0] (1996)
Droloxifène [82413-20-5] (1996)
Dulcine [150-69-6] (1987)
Eau potable chlorée (1991)
Eclairage fluorescent (1992)
Endrine [72-20-8] (1987)
Eosine [15086-94-9] (1987)
Epithioéthane [420-12-2] (1987)
3,4-Epoxy-6-méthylcyclohexylméthyl-3,4-époxy-6-méthyl-cyclohexane carboxylate [141-37-7] (1999)
Estazolam [29975-16-4] (1996)
Ethionamide [536-33-4] (1987)
Ethylène [74-85-1] (1994)
Eugénol [97-53-0] (1987)
Fenvalérate [51630-58-1] (1991)
Ferbam [14484-64-1] (1987)
Fibres acryliques (1987)
Fibres modacryliques (1987)
Fibrilles de p-aramide [24938-64-5] (1997)
Filaments de verre (1988)
Fluométuron [2164-17-2] (1987)
Fluoranthène [206-44-0] (1987)
Fluorène [86-73-7] (1987)
5-Fluorouracile [51-21-8] (1987)
Fluorure de vinylidène [75-38-7] (1999)
Fluorures (inorganiques, utilisés dans l’eau potable) (1987)
Furazolidone [67-45-8] (1987)
Furfural [98-01-1] (1995)
Furosémide (Frusémide) [54-31-9] (1990)
Gemfibrozil [25812-30-0] (1996)
Gyromitrine [16568-02-8] (1987)
Hématite [1317-60-8] (1987)
Hexachlorobutadiène [87-68-3] (1999)
Hexachlorophène [70-30-4] (1987)
Huiles isopropyliques (1999)
Hydralazine [86-54-4] (1987)
Hydrate de chloral [302-17-0] (1995)
Hydrazide de l’acide isonicotinique (Isoniazide) [54-85-3] (1987)
Hydrazide maléique [123-33-1] (1987)
Hydrochlorothiazide [58-93-5] (1990)
Hydroquinone [123-31-9] (1999)
4-Hydroxyazobenzène [1689-82-3] (1987)
8-Hydroxyquinoléine [148-24-3] (1987)
8-Hydroxyquinoléine de cuivre [10380-28-6] (1987)
Hydroxysenkirkine [26782-43-4] (1987)
Hydroxytoluène butylé (BHT) [128-37-0] (1987)
Hypochlorites (1991)
Iodure de méthyle [74-88-4] (1999)
Isatidine [15503-86-3] (1987)
Isocyanate d’allyle [57-06-7] (1999)
Isophosphamide [3778-73-2] (1987)
Isopropanol [67-63-0] (1999)
Isosafrol [120-58-1] (1987)
Isothiocyanate d’allyle [57-06-7] (1999)
Isovalérate d’allyle [2835-39-4] (1999)
Jacobine [6870-67-3] (1987)
Jaune AB [85-84-7] (1987)
Jaune dispersé 3 [2832-40-8] (1990)
Jaune HC 4 [59820-43-8] (1993)
Jaune OB [131-79-3] (1987)
Jaune Sunset FCF [2783-94-0] (1987)
Jaune Vat 4 [128-66-5] (1990)
Kaempférol [520-18-3] (1987)
d-Limonène [5989-27-5] (1999)10
Lutéoskyrine [21884-44-6] (1987)
Malathion [121-75-5] (1987)
Malonaldéhyde [542-78-9] (1999)
Manèbe [12427-38-2] (1987)
Medphalane [13045-94-8] (1987)
Mélamine [108-78-1] (1999)10
6-Mercaptopurine [50-44-2] (1987)
Mercure [7439-97-6] et composés du mercure inorganique (1993)
Mésylate d’hycanthone [23255-93-8] (1987)
Métabisulfites (1992)
Méthacrylate de méthyle [80-62-6] (1994)
Méthotrexate [59-05-2] (1987)
Méthoxychlore [72-43-5] (1987)
5-Méthylangélicine [73459-03-7] et exposition aux rayonnements ultraviolets A (1987)
Méthyl-tert-butyléther [1634-04-4] (1999)
1-Méthylchrysène [3351-28-8] (1987)
2-Méthylchrysène [3351-32-4] (1987)
3-Méthylchrysène [3351-31-3] (1987)
4-Méthylchrysène [3351-30-2] (1987)
6-Méthylchrysène [1705-85-7] (1987)
N-Méthyl-N,4-dinitrosoaniline [99-80-9] (1987)
4,4´-Méthylènebis (N,N-diméthylaniline) [101-61-1] (1987)
4-4´-Méthylènediphényl diisocyanate [101-68-8] (1999)
2-Méthylfluoranthène [33543-31-6] (1987)
3-Méthylfluoranthène [1706-01-0] (1987)
Méthylglyoxal [78-98-8] (1991)
N-Méthylolacrylamide [90456-67-0] (1994)
Méthylparathion [298-00-0] (1987)
1-Méthylphénanthrène [832-69-9] (1987)
7-Méthylpirido[3,4-c]psoralène [85878-63-3] (1987)
Monuron [150-68-5] (1991)
Morpholine [110-91-8] (1999)
Mousses de polyuréthane [9009-54-5] (1987)
Moutarde d’œstradiol [22966-79-6] (1987)
Musc ambrette [83-66-9] (1996)
Musc xylène [81-15-2] (1996)
1,5-Naphtalène diisocyanate [3173-72-6] (1999)
1-Naphtylamine [134-32-7] (1987)
1-Naphtylthiourée (ANTU) [86-88-4] (1987)
Nithiazide [139-94-6] (1987)
5-Nitro-o-anisidine [99-59-2] (1987)
9-Nitroanthracène [602-60-8] (1987)
7-Nitrobenzo[a]anthracène [20268-51-3] (1989)
6-Nitrobenzo[a]pyrène [63041-90-7] (1989)
4-Nitrobiphényle [92-93-3] (1987)
3-Nitrofluoranthène [892-21-7] (1987)
Nitrofural (Nitrofurazone) [59-87-0] (1990)
Nitrofurantoïne [67-20-9] (1990)
1-Nitronaphtalène [86-57-7] (1989)
2-Nitronaphtalène [581-89-5] (1989)
3-Nitropérylène [20589-63-3] (1989)
2-Nitropyrène [789-07-1] (1989)
N´-Nitrosoanabasine [37620-20-5] (1987)
N-Nitrosoanatabine [71267-22-6] (1987)
N-Nitrosodiphénylamine [86-30-6] (1987)
p-Nitrosodiphénylamine [156-10-5] (1987)
N-Nitrosoguvacine [55557-01-2] (1987)
N-Nitrosoguvacoline [55557-02-3] (1987)
N-Nitrosohydroxyproline [30310-80-6] (1987)
3-(N-Nitrosométhylamino)propionaldéhyde [85502-23-4] (1987)
4-(N-Nitrosométhylamino)-4-(3-pyridyl)-1-butanal (NNA) [64091-90-3] (1987)
N-Nitrosoproline [7519-36-0] (1987)
Nitrotoluènes (mélange d’isomères) [88-72-2; 99-08-1; 99-99-0] (1996)
5-Nitro-o-toluidine [99-55-8] (1990)
Nitrovine [804-36-4] (1987)
Nylon 6 [25038-54-4] (1987)
Oléate de glycidyle [5431-33-4] (1987)
Opisthorchis felineus (infection) (1994)
Orangé acide CI-3 [6373-74-6] (1993)
Orangé d’acridine [494-38-2] (1987)
Orangé G [1936-15-8] (1987)
Orangé I [523-44-4] (1987)
Oxyde de décabromodiphényle [1163-19-5] (1999)
Oxyde de fer saccharique [8047-67-4] (1987)
Oxyde de tris(1-aziridinyl)phosphine [545-55-1] (1987)
Oxyde de tris(2-méthyl-1-aziridinyl)phosphine [57-39-6] (1987)
Oxyde ferrique (III) [1309-37-1] (1987)
Oxyphenbutazone [129-20-4] (1987)
Palygorskite (attapulgite) [12174-11-7] (fibres courtes, < 5 µm) (1997)
Paracétamol (Acétaminophène) [103-90-2] (1999)
Parathion [56-38-2] (1987)
Patuline [149-29-1] (1987)
Pentachloroéthane [76-01-7] (1999)
Perméthrine [52645-53-1] (1991)
Peroxyde de benzoyle [94-36-0] (1999)
Peroxyde de lauroyle [105-74-8] (1999)
Peroxyde d’hydrogène [7722-84-1] (1999)
Pérylène [198-55-0] (1987)
Pétasiténine [60102-37-6] (1987)
Phénanthrène [85-01-8] (1987)
Phénicarbazide [103-03-7] (1987)
Phénol [108-95-2] (1999)
Phénylbutazone [50-33-9] (1987)
m-Phénylènediamine [108-45-2] (1987)
p-Phénylènediamine [106-50-3] (1987)
N-Phényl-2-naphtylamine [135-88-6] (1987)
o-Phénylphénol [90-43-7] (1999)
Phosphate de tris(2-chloréthyle) [115-96-8] (1999)
Phosphite acide de diméthyle [868-85-9] (1999)
Phtalate de butylbenzyle [85-68-7] (1999)
Picloram [1918-02-1] (1991)
Pirido [3,4-c]psoralène [85878-62-2] (1987)
Poly(acétate de vinyle) [9003-20-7] (1987)
Poly(alcool vinylique) [9002-89-5] (1987)
Polychloroprène [9010-98-4] (1987)
Poly(chlorure de vinyle) [9002-86-2] (1987)
Polyéthène [9002-88-4] (1987)
Poly(méthacrylate de méthyle) [9011-14-7] (1987)
Polyméthylène polyphényle isocyanate [9016-87-9] (1987)
Polypropylène [9003-07-0] (1987)
Polystyrène [9003-53-6] (1987)
Polytétrafluoroéthylène [9002-84-0] (1987)
Polyvinylpyrrolidone [9003-39-8] (1999)
Ponceau SX [4548-53-2] (1987)
Potassium bis(2-hydroxyéthyle)dithiocarbamate [23746-34-1] (1987)
Poussières de charbon (1997)
Prazépam [2955-38-6] (1996)
Prednimustine [29069-24-7] (1990)
Prednisone [53-03-2] (1987)
Propham [122-42-9] (1987)
n-Propyle carbamate [627-12-3] (1987)
Propylène [115-07-1] (1994)
Ptaquiloside [87625-62-5] (1987)
Pyrène [129-00-0] (1987)
Pyriméthamine [58-14-0] (1987)
Quercétine [117-39-5] (1999)
p-Quinone [106-51-4] (1999)
Quintozène (Pentachloronitrobenzène) [82-68-8] (1987)
Réserpine [50-55-5] (1987)
Résorcinol [108-46-3] (1999)
Rétrorsine [480-54-6] (1987)
Rhodamine B [81-88-9] (1987)
Rhodamine 6G [989-38-8] (1987)
Riddelliine [23246-96-0] (1987)
Rifampicine [13292-46-1] (1987)
Ripazépam [26308-28-1] (1996)
Rouge D et C 9 [5160-02-1] (1993)
Rouge de méthyle [493-52-7] (1987)
Rouge écarlate [85-83-6] (1987)
Rouge HC 3 [2871-01-4] (1993)
Rouge pigment CI-3 [2425-85-6] (1993)
Rouge Soudan 7B [6368-72-5] (1987)
Rugulosine [23537-16-8] (1987)
Saccharine [81-07-2] et ses sels (1999)11
Schistosoma mansoni (infection) (1994)
Sélénium [7782-49-2] et composés du sélénium (1987)
Sels de proflavine (1987)
Sels de tétrakis (hydroxyméthyl) phosphonium (1999)
Sénéciphylline [480-81-9] (1987)
Senkirkine [2318-18-5] (1987)
Sépiolite [15501-74-3] (1997)
Silice amorphe [7631-86-9] (1997)
Simazine [122-34-9] (1999)
Soudan I [842-07-9] (1987)
Soudan II [3118-97-6] (1987)
Soudan III [85-86-9] (1987)
Spironolactone [52-01-7] (1987)
Stéarate de glycidyle [7460-84-6] (1987)
Sulfafurazole (Sulfisoxazole) [127-69-5] (1987)
Sulfaméthoxazole [723-46-6] (1987)
Sulfate de phénelzine [156-51-4] (1987)
Sulfate de vinblastine [143-67-9] (1987)
Sulfate de vincristine [2068-78-2] (1987)
Sulfites (1992)
Sulfure de bis(1-aziridinyl)morpholinophosphine [2168-68-5] (1987)
Symphytine [22571-95-5] (1987)
Talc [14807-96-6] sans fibres asbestiformes (1987)
Témazépam [846-50-4] (1996)
2,2´ ,5,5´-Tétrachlorobenzidine [15721-02-5] (1987)
1,1,1,2-Tétrachloroéthane [630-20-6] (1999)
1,1,2,2-Tétrachloroéthane [79-34-5] (1999)
Tétrachlorvinphos [22248-79-9] (1987)
Théobromine [83-67-0] (1991)
Théophylline [58-55-9] (1991)
Thiouracile [141-90-2] (1987)
Thiram [137-26-8] (1991)
Toluène [108-88-3] (1999)
Torémifène [89778-26-7] (1996)
Toxines dérivées du Fusarium graminearum, du F. culmorum et du F. crookwellense (1993)
Toxines dérivées du Fusarium sporotrichioides (1993)
Trichlorfon [52-68-6] (1987)
Trichloroacétonitrile [545-06-2] (1999)
1,1,1-Trichloroéthane [71-55-6] (1999)
1,1,2-Trichloroéthane [79-00-5] (1999)
Triéthylèneglycol diglydicyléther [1954-28-5] (1999)
Trifluraline [1582-09-8] (1991)
4,4´,6-Triméthylangélicine [90370-29-9] et exposition aux rayonnements ultraviolets A (1987)
2,4,5-Triméthylaniline [137-17-7] (1987)
2,4,6-Triméthylaniline [88-05-1] (1987)
4,5´ ,8-Triméthylpsoralène [3902-71-4] (1987)
2,4,6-Trinitrotoluène [118-96-7] (1996)
Triphénylène [217-59-4] (1987)
Tris(aziridinyl)-p-benzoquinone (Triaziquone) [68-76-8] (1987)
2,4,6-Tris(1-aziridinyl)-s-triazine [51-18-3] (1987)
1,2,3-Tris-(chlorométhoxy)propane [38571-73-2] (1999)
Trisulfure d’antimoine [1345-04-6] (1989)
Vert Guinée B [4680-78-8] (1987)
Vert intense FCF [2353-45-9] (1987)
Vert lumière SF [5141-20-8] (1987)
N-Vinyl-2-pyrrolidone [88-12-0] (1999)
Vinyltoluène [25013-15-4] (1994)
Virus de l’hépatite D (1994)
Virus humain de la leucémie à cellules T, type II (1996)
Wollastonite [13983-17-0] (1997)
Xylène [1330-20-7] (1999)
2,4-Xylidine [95-68-1] (1987)
2,5-Xylidine [95-78-3] (1987)
Zectrane [315-18-4] (1987)
Zéolites [1318-02-1] autres que érionite (clinoptilolite, phillipsite, mordénite, zéolite non fibreux japonais, zéolites synthétiques) (1997)
Zinèbe [12122-67-7] (1987)
Ziram [137-30-4] (1991)
Bitumes [8052-42-4] raffinés à la vapeur ou à l’air, résidus du crackage (1987)
Carburants diesel, distillat (léger) (1989)
Carburéacteur (1989)
Encres d’imprimerie (1996)
Fuels, distillat (léger) (1989)
Huiles minérales, hautement raffinées (1987)
Mastication de bétel sans tabac (1987)
Maté (1991)
Pétrole brut [8002-05-9] (1989)
Solvants de pétrole (1989)
Terpènes polychlorés (Strobane®) [8001-50-1] (1987)
Thé (1991)
Articles en cuir (fabrication) (1987)
Bois d’œuvre et bois de sciage (industries) (y compris exploitation du bois) (1987)
Colorants capillaires (usage personnel) (1993)
Papier et de pâte à papier (fabrication) (1987)
Peinture (fabrication) (exposition professionnelle) (1989)
Tannage et traitement du cuir (1987)
Verre ordinaire et verres spéciaux (fabrication) (1993)
Caprolactame [105-60-2] (1998)
1 Lorsqu'il y a lieu, le numéro CAS (Chemical Abstracts Registry) figure entre crochets; l'année indiquée entre parenthèses correspond à l'année de publication de l'évaluation la plus récente (pour plus de détails, consultez la monographie pertinente (publiée en anglais seulement)).
2 Cette évaluation s'applique à l'ensemble du groupe, mais pas nécessairement à chacun des agents du groupe.
3 Evalués en groupe.
4 On dispose également d'indications qui permettent de conclure que ces agents jouent un rôle protecteur contre les cancers de l'ovaire et de l'endomètre.
5 On dispose également d'indications qui permettent de conclure que cet agent réduit le risque de cancer du sein controlatéral.
6 Modification de l'évaluation globale, du groupe 2A au groupe 1, sur la base de données complémentaires relatives à l'évaluation de la cancérogénicité et à ses mécanismes.
7 Modification de l'évaluation globale, du groupe 2B au groupe 2A, sur la base de données complémentaires relatives à l'évaluation de la cancérogénicité et à ses mécanismes.
8 Modifications de l'évaluation globale, du groupe 3 au groupe 2B, sur la base de données complémentaires relatives à l'évaluation de la cancérogénicité et à ses mécanismes.
9 Il existe certaines indications selon lesquelles le risque de cancer du côlon serait inversement proportionnel à la consommation de café; il n'a pas été possible de classer la consommation de café quant à sa cancérogénicité pour d'autres organes.
10 Données complémentaires relatives à l'évaluation de la cancérogénicité et à ses mécanismes prises en complte dans l'évaluation globale.
11 Modification de l'évaluation globale, du groupe 2B au groupe 3 sut la case de données complémentaires relatives à l'évaluation de la cancérogénicité et à ses mécanismes.
Les principes et les méthodes employés pour évaluer le risque que présentent les produits chimiques non cancérogènes sont semblables dans les différentes parties du monde, mais il est frappant de constater combien ces approches sont disparates dans le cas des agents chimiques cancérogènes. D’un pays à l’autre, on constate des différences marquées et, dans un même pays, les organismes de réglementation, les comités et les scientifiques spécialisés dans l’évaluation du risque appliquent ou recommandent d’appliquer des démarches différentes. L’évaluation du risque pour les substances non cancérogènes est une pratique relativement bien établie et assez cohérente, notamment parce que — contrairement à ce qui se passe dans le cas des agents cancérogènes — on l’applique depuis longtemps et parce qu’on connaît bien leur toxicité; de plus, les méthodes utilisées et les informations qu’on en tire font la quasi-unanimité chez les scientifiques et dans l’opinion publique et leur inspirent confiance.
Pour pallier les incertitudes dont souffrent les données toxicologiques sur les agents non cancérogènes (obtenues essentiellement à partir d’expériences animales) et permettre leur application à grande échelle à des populations humaines hétérogènes, on a introduit des facteurs de sécurité (appelés aussi coefficients de sécurité). On a ensuite fixé — grâce à cette méthode du facteur de sécurité ou d’incertitude — des valeurs limites recommandées ou obligatoires qui assurent à l’être humain une exposition dépourvue de danger et qui correspondaient en général à une fraction de la dose d’exposition chez l’animal ne comportant aucun effet nocif observable (NOAEL) ou de la plus faible dose (ou première dose) induisant un effet nocif observable (LOAEL). On estimait que tant que l’exposition humaine n’excédait pas ces limites recommandées, les substances chimiques dangereuses ne pouvaient avoir d’effets nocifs. On applique toujours la même technique, sous une forme plus affinée, pour évaluer le risque toxique de nombreux produits chimiques.
De la fin des années soixante au début des années soixante-dix, les organismes réglementaires, aux Etats-Unis tout d’abord, ont été confrontés à un problème de plus en plus préoccupant que de nombreux scientifiques estimaient ne pouvoir résoudre grâce à l’approche basée sur un facteur de sécurité. Ils allaient même jusqu’à penser que cette technique était inadaptée, pour ne pas dire dangereuse. En effet, il existe des produits chimiques qui, dans des conditions bien précises, font augmenter le risque de cancer chez l’humain ou l’animal. Pour des raisons pratiques, ces substances ont été rattachées aux cancérogènes. De nos jours encore, la définition des produits cancérogènes fait l’objet de débats et de controverses et les avis divergent quant à la façon de les identifier et de les classer; il en est de même pour les mécanismes d’induction du cancer par les produits chimiques.
Ce débat avait débuté bien avant, lorsque les scientifiques ont découvert dans les années quarante que les cancérogènes chimiques déterminent des lésions par un mécanisme biologique d’un genre totalement différent des autres formes de toxicité. Partant des principes de la biologie des cancers induits par les rayonnements, ces scientifiques ont avancé l’hypothèse de l’existence d’un «non-seuil» applicable à la fois aux rayonnements et aux agents chimiques cancérogènes. Pour eux, toute exposition à un agent cancérogène augmente la probabilité (le risque) de développer un cancer, dès lors que le produit atteint sa cible biologique critique, le matériel génétique en particulier, et interagit avec elle.
Parallèlement à ce débat scientifique sur les seuils, on a vu le public se préoccuper chaque jour un peu plus du risque chimique cancérogène et vouloir sans délai se protéger des diverses pathologies rassemblées sous le terme de cancer. Le cancer, avec son caractère insidieux, sa longue période de latence et sa tendance à augmenter dans la population, était considéré par le grand public et par les politiciens comme une affaire sérieuse qui méritait la plus grande attention. Les instances réglementaires se trouvaient donc confrontées à une situation où un grand nombre de personnes, parfois la presque totalité d’une population, étaient ou pouvaient être exposées à des concentrations relativement faibles de substances chimiques (dans les produits de consommation, les médicaments, sur le lieu de travail ou encore dans l’air, l’eau, la nourriture et le sol) dont on avait établi le pouvoir cancérogène chez l’humain ou chez l’animal de laboratoire après des expositions à des concentrations relativement élevées.
Ces responsables de la réglementation devaient donc faire face à deux questions fondamentales auxquelles on ne pouvait répondre de manière satisfaisante compte tenu des connaissances scientifiques de l’époque:
Les autorités réglementaires ont admis qu’il était nécessaire de pouvoir s’appuyer sur des hypothèses établies parfois sur des bases scientifiques, mais souvent aussi en l’absence de preuve expérimentale. Dans un but de cohérence, des définitions et une série d’hypothèses ont donc été élaborées en vue d’une application à l’ensemble des produits cancérogènes.
De nombreux arguments tendent à prouver que la cancérogenèse chimique est un processus à étapes multiples sous la dépendance de lésions génétiques et épigénétiques. Cette théorie est avalisée par bien des membres de la communauté scientifique à travers le monde (Barrett, 1993). On a pris l’habitude de distinguer trois étapes dans le processus de cancérogenèse chimique: l’initiation, la promotion et la progression, mais on ne connaît pas le nombre exact des modifications génétiques impliquées dans ce processus.
L’initiation suppose l’induction d’une modification cellulaire irréversible; pour les cancérogènes génotoxiques, cette modification est toujours assimilée à un événement mutationnel. Theodor Boveri, dont bien des hypothèses et des prédictions se sont révélées exactes par la suite, soupçonnait déjà en 1914 que la mutagenèse était un mécanisme de la cancérogenèse. Du fait que la plus petite quantité d’un cancérogène modifiant l’ADN peut provoquer des mutations irréversibles et autoréplicatives, on estime qu’il n’existe aucun seuil. La promotion est le processus par lequel une cellule initiée se développe (formation d’un clone) grâce à une série de divisions et forme des lésions (pré)néoplasiques. De nombreux débats ont lieu pour savoir si les cellules initiées subissent d’autres modifications génétiques au cours de cette phase de transition.
Enfin, lors de l’étape de progression, l’«immortalité» est atteinte et des tumeurs malignes peuvent se développer en induisant une angiogenèse et en échappant aux systèmes de contrôle de l’hôte. Cette étape est caractérisée par une croissance invasive et fréquemment par une propagation métastasique de la tumeur. La progression s’accompagne d’autres modifications génétiques du fait de l’instabilité des cellules en prolifération et de la sélection.
On distingue donc trois mécanismes généraux par lesquels une substance peut influencer ce processus cancérogène multiétapes. Un produit chimique peut induire une lésion génétique déterminante, promouvoir ou faciliter l’expansion clonale d’une cellule initiée ou encore stimuler la progression vers la malignité par des modifications somatiques ou génétiques.
On peut dire du risque qu’il s’agit de la fréquence de survenue, prévue ou réelle, d’un effet nocif pour l’humain ou l’environnement, par suite de l’exposition à un danger. L’évaluation du risque est une méthode d’organisation systématique de l’information scientifique avec ses incertitudes pour décrire et qualifier les risques que des substances, des processus, des actions ou des événements dangereux présentent pour la santé. Elle nécessite une évaluation des informations pertinentes et la sélection de modèles permettant d’en tirer des conclusions. De plus, elle requiert la prise en considération des incertitudes, tout en gardant à l’esprit qu’une interprétation différente des données disponibles reste plausible du point de vue scientifique. La terminologie utilisée actuellement pour l’évaluation du risque a été proposée en 1984 par l’Académie nationale des sciences des Etats-Unis (NAS). L’évaluation qualitative du risque est devenue la caractérisation ou l’identification du danger, et l’évaluation quantitative du risque a été scindée en trois: relation dose-réponse, évaluation de l’exposition et caractérisation du risque.
Dans la rubrique suivante, nous abordons ces différents aspects en nous fondant sur nos connaissances actuelles du processus de cancérogenèse (chimique). Il apparaîtra clairement que la principale incertitude pour évaluer un risque cancérogène concerne la relation dose-réponse aux faibles concentrations telles qu’on les rencontre lors d’une exposition environnementale.
Ce processus consiste à rechercher les produits susceptibles de provoquer un cancer chez l’humain ou, si l’on veut, à déterminer leurs propriétés génotoxiques intrinsèques. Les agents cancérogènes sont classés sur la base de leurs propriétés et d’informations d’origine diverse et notamment:
Pour identifier un danger, il faut classer les produits chimiques en groupes en se basant sur leur caractère cancérogène chez l’animal, ou sur des données épidémiologiques dans l’espèce humaine lorsqu’elles sont disponibles. Les méthodes de classification des agents chimiques cancérogènes les plus connues sont celles du CIRC (1987), de l’Union européenne (1991) et de l’Agence américaine de protection de l’environnement (EPA) (1986). Le tableau 33.17 résume les critères utilisés pour le classement (en particulier les méthodes d’extrapolation aux faibles doses).
|
|
EPA (Etats-Unis) actuel |
Danemark |
CEE |
Royaume-Uni |
Pays-Bas |
Norvège |
|
Cancérogène génotoxique |
Processus multiétapes linéarisés utilisant le modèle le plus approprié |
MLE à partir des modèles 1- et 2-étapes avec estimation du meilleur résultat |
Aucune procédure spécifiée |
Aucun modèle, expertise scientifique et jugement à partir de toutes les données valables |
Modèle linéaire utilisant la DT50 (méthode Peto) ou �Méthode néerlandaise simple� en l�absence de DT50 |
Aucune procédure spécifiée |
|
Cancérogène non génotoxique |
Idem |
Modèle biologique de Thorslund ou modèle multiétapes ou modèle de Mantel-Bryan, basé sur l�origine tumorale et la relation dose-réponse |
Utilisation du NOAEL et de facteurs de sécurité |
Utilisation du NOEL et de facteurs de sécurité pour déterminer la dose journalière admissible |
Utilisation du NOEL et de facteurs de sécurité pour déterminer la dose journalière admissible |
|
La classification des agents cancérogènes soulève un problème important, dont les conséquences sont parfois déterminantes pour la réglementation: celui de la distinction entre les mécanismes d’action génotoxiques et non génotoxiques. L’hypothèse par défaut de l’EPA aux Etats-Unis est qu’il n’existe pas de seuil pour les substances ayant une activité cancérogène chez les animaux de laboratoire (ou du moins l’existence d’un seuil ne peut être démontrée): quelle que soit l’exposition, le risque est toujours présent. Cette position correspond à ce que l’on a pris l’habitude d’appeler l’hypothèse de non-seuil ou d’absence de seuil pour les composés génotoxiques (produisant des lésions de l’ADN). L’Union européenne et bon nombre de ses Etats membres, comme le Danemark, les Pays-Bas et le Royaume-Uni, font une distinction entre les cancérogènes génotoxiques et ceux qu’on soupçonne de produire des tumeurs par des mécanismes non génotoxiques. Pour les agents cancérogènes génotoxiques, les procédures d’évaluation quantitative dose-réponse supposent également l’absence de seuil, bien que les procédures diffèrent de celles utilisées par l’EPA. Pour les substances non génotoxiques, on suppose qu’il existe un seuil, et les procédures dose-réponse utilisées en supposent l’existence. Pour ces substances, comme pour les produits non cancérogènes, l’évaluation du risque est généralement effectuée en appliquant un facteur de sécurité.
Il est utile de rappeler que les procédures d’évaluation du risque ont été développées dans un contexte et un cadre différents. Celles du CIRC n’ont pas été proposées dans un but réglementaire, bien qu’on s’en soit servi pour établir des lignes directrices à visée réglementaire. La procédure de l’EPA a été conçue comme une base de décision pour élaborer une évaluation quantitative du risque, alors que celle de l’Union européenne est utilisée actuellement aux fins de l’étiquetage des produits chimiques (symbole classe de risque et phrase décrivant le risque). Moolenaar a publié (Moolenaar, 1994) une synthèse bibliographique de toutes les méthodes employées par huit agences gouvernementales et deux organismes indépendants souvent cités: le Centre international de recherche sur le cancer (CIRC) et la Conférence américaine des hygiénistes gouvernementaux du travail (ACGIH).
Les procédures de classification ne prennent généralement pas en compte l’ensemble des éléments négatifs disponibles. Depuis quelques années, on commence à mieux comprendre le mécanisme d’action des cancérogènes. On sait maintenant que certains de ces mécanismes sont spécifiques à des espèces particulières et ne se retrouvent pas chez l’être humain. Il faut citer ici deux exemples pour illustrer le propos. Premièrement, des études récentes sur le pouvoir cancérogène des particules de carburant diesel ont montré que le rat développe des tumeurs pulmonaires en réponse à l’accumulation de ces particules dans les poumons, alors qu’on n’a jamais observé de cancer pulmonaire chez les mineurs de charbon ayant une charge importante à ce niveau. Deuxièmement, les tumeurs rénales observées chez le rat mâle ne sont pas considérées comme pertinentes du fait qu’elles résultent de l’accumulation rénale de l’α-2 microglobuline, protéine qui n’existe pas dans l’espèce humaine (Borghoff, Short et Swenberg, 1990). D’autres exemples de ce type peuvent être mentionnés: perturbations de la fonction thyroïdienne ou de la prolifération des peroxysomes chez les rongeurs, ou anomalies de la mitose au niveau hépatique chez la souris.
Ces notions permettent une interprétation plus perspicace des études de cancérogenèse. On doit encourager les recherches conduisant à une meilleure compréhension des mécanismes d’action de la cancérogenèse, car elles permettront d’améliorer la classification des produits et d’ajouter une catégorie où pourront être classés les produits chimiques non cancérogènes pour l’humain.
Dans l’évaluation de l’exposition, on considère souvent que l’évaluation du risque est le facteur le moins incertain pour deux raisons: parce qu’il est possible dans certains cas de contrôler les expositions et parce qu’il existe des modèles d’exposition assez bien validés. Ce n’est vrai qu’en partie puisque la plupart des évaluations d’exposition ne tirent pas suffisamment parti de l’ensemble des informations disponibles. Il reste donc encore beaucoup à faire pour améliorer l’évaluation de l’exposition, qu’elle soit externe ou interne. Dans le cas des agents cancérogènes, en particulier, l’établissement des relations dose-réponse à partir des concentrations atteintes au niveau des cibles tissulaires plutôt qu’à partir des niveaux d’exposition extérieure devrait conduire à une meilleure prévision du risque, bien qu’on soit alors appelé à faire de nombreuses hypothèses par défaut. Les modèles pharmacocinétiques basés sur la physiologie permettant de déterminer la concentration des métabolites réactifs au niveau des tissus cibles revêtent à cet égard un très grand intérêt.
La dose ou le niveau d’exposition responsable d’un effet dans une étude chez l’animal et la dose susceptible de produire un effet semblable dans l’espèce humaine sont des paramètres essentiels à la caractérisation du risque. Ces paramètres incluent à la fois l’évaluation de la relation dose-réponse depuis les doses élevées jusqu’aux faibles doses et l’extrapolation interespèces. L’extrapolation pose un problème de logique: les données sont extrapolées de plusieurs ordres de grandeur au-dessous des taux d’exposition expérimentaux au moyen de modèles empiriques qui ne reflètent pas les mécanismes sous-jacents de la cancérogenèse. Cette manière de procéder enfreint donc un principe de base lors de l’application d’un modèle empirique, selon lequel on ne doit pas extrapoler en dehors de la gamme des données observées. Par conséquent, cette extrapolation empirique entraîne un degré d’incertitude important, tant du point de vue statistique que du point de vue biologique. Actuellement, aucun modèle mathématique n’est reconnu comme étant le plus adapté à l’extrapolation aux faibles doses en cancérogenèse. Les modèles mathématiques utilisés pour définir la relation existant entre la dose externe administrée, le temps et la survenue d’une tumeur sont basés sur des hypothèses probabilistes ou mécanistiques, parfois sur les deux. On trouve au tableau 33.18 une liste des modèles les plus fréquemment cités (Kramer et coll., 1995).
|
Modèles probabilistes |
Modèles mécanistiques |
|
|
|
Modèles par atteintes |
Modèles biologiques |
|
Logit |
Modèle monoatteinte |
Modèles temps/tumeur (MVK)1 |
|
Probit |
Modèle atteintes multiples |
Cohen et Ellwein |
|
Mantel-Bryan |
Weibull (Pike)1 |
|
|
Weibull |
Modèle étapes multiples (Armitage-Doll)1 |
|
|
Modèle Gamma atteintes multiples |
Modèle multiétapes linéarisé |
|
1 MVK = Moolgavkar-Venzon-Knudson.
Ces modèles dose-réponse sont en général appliqués à des études de cancérogenèse effectuées selon un protocole standard comportant un nombre limité de doses expérimentales. Au lieu d’établir la courbe dose-réponse complète, une étude du pouvoir cancérogène est en général limitée à trois (ou deux) doses relativement fortes, la dose la plus élevée correspondant à la dose maximale admissible. L’utilisation de fortes doses permet de surmonter la faible sensibilité statistique (10 à 15% au-dessus du bruit de fond) inhérente à de telles études, due (notamment pour des raisons pratiques) au nombre relativement restreint d’animaux utilisés. Etant donné l’absence de résultats dans la zone des faibles doses (ils ne sont pas déterminés expérimentalement), il est nécessaire d’extrapoler au-delà de la gamme d’observation. Pour la plupart des résultats, les modèles mentionnés ci-dessus conviennent tous bien à la gamme des doses étudiées, en raison du nombre limité de doses et d’animaux. Néanmoins, dans la zone des faibles doses, ces modèles divergent de plusieurs ordres de grandeur, ce qui introduit une marge importante d’incertitude dans l’estimation du risque pour de tels niveaux d’exposition.
La forme réelle de la courbe dose-réponse dans la gamme des faibles doses ne pouvant être obtenue expérimentalement, il est indispensable de connaître le mécanisme de la cancérogenèse si l’on veut pouvoir choisir à bon escient le modèle qui convient le mieux. Kramer et coll. (1995) et Park et Hawkins (1993) ont effectué des synthèses bibliographiques détaillées sur les divers aspects des modèles d’extrapolation mathématiques.
A côté des modèles mathématiques utilisés de nos jours, plusieurs autres approches ont été proposées récemment.
Actuellement, les modèles biologiques tels que le modèle de Moolgavkar-Venzon-Knudson (MVK) sont très prometteurs, mais ils ne sont pas encore suffisamment évolués pour une utilisation en routine et nécessitent des informations spécifiques que ne peuvent fournir les études expérimentales. Des études très poussées (sur 4 000 rats) comme celles réalisées avec les N-nitrosoalkylamines donnent une idée de la dimension des travaux nécessaires au recueil de ces informations. Cependant, ces études ne permettent toujours pas une extrapolation aux faibles doses. Tant que ces modèles ne seront pas plus élaborés, ils ne pourront être utilisés que pour des applications ponctuelles.
L’utilisation de modèles mathématiques pour extrapoler en dessous de la gamme des doses expérimentales est en fait l’équivalent d’une approche par facteur de sécurité, avec un facteur d’incertitude important et mal défini. L’alternative la plus simple serait d’appliquer un facteur d’évaluation au «niveau sans effet observé» ou au «plus faible niveau testé». Le niveau utilisé pour ce facteur d’évaluation devrait être déterminé cas par cas en considérant la nature du produit chimique et la population exposée.
Cette approche, basée sur un modèle mathématique adapté aux données expérimentales à l’intérieur de la gamme d’observation, est employé pour estimer ou interpoler une dose correspondant à un niveau donné d’effet, tel que 1%, 5% ou 10% d’augmentation d’incidence tumorale (DE01, DE05, DE10). Une augmentation de 10% correspondant au plus petit changement pouvant être déterminé statistiquement dans une étude expérimentale standard, la DE10 convient donc bien aux données de cancérogenèse. Le fait d’utiliser une dose de référence qui se trouve à l’intérieur de la gamme d’observation expérimentale évite les problèmes que peut poser l’extrapolation de la dose. La dose de référence ou sa limite de confiance inférieure reflètent les doses auxquelles surviennent des changements d’incidence tumorale et sont totalement indépendantes du modèle mathématique utilisé. On peut employer la dose de référence comme mesure du potentiel tumoral dans l’évaluation du risque et, en l’associant à des facteurs d’évaluation appropriés, s’en servir pour fixer des niveaux admissibles pour une exposition humaine.
Krewski et coll. (1990) ont effectué une synthèse bibliographique des études sur le seuil de réglementation pour les produits chimiques cancérogènes. Ils ont constaté, à partir des résultats de 585 expériences sur le potentiel cancérogène, que la dose correspondant au niveau de risque 10–6 a une distribution approximativement log-normale autour d’une médiane de 70 à 90 ng/kg/jour. Toute exposition à des doses supérieures doit donc être considérée comme inacceptable. Cette dose est obtenue par extrapolation linéaire à partir de la DT50 (dose toxique pour 50% des animaux traités) et se trouve dans les limites d’un facteur de cinq à dix par rapport au résultat que donne le modèle multiétapes linéarisé. Cependant, les valeurs de la TD50 sont reliées à la dose maximale admissible, ce qui jette un doute sur la validité de la mesure. En dépit de cela, la DT50 est souvent très proche ou même à l’intérieur de la gamme des données expérimentales.
Avant de pouvoir envisager l’utilisation d’un tel seuil de réglementation, il serait nécessaire de tenir davantage compte des données biologiques, analytiques et mathématiques et de disposer d’une base de données beaucoup plus fournie. Des recherches complémentaires sur le pouvoir de divers agents cancérogènes permettront d’apporter un meilleur éclairage dans ce domaine.
Si l’on considère les espoirs qui ont été à l’origine de la réglementation sur les produits cancérogènes (de l’environnement), essentiellement une réduction sensible du nombre de cancers, les résultats sont plutôt décevants. Au fil des ans, on s’est aperçu que le nombre des cas de cancers attribués à des cancérogènes réglementés était étonnamment faible. Malgré les efforts de réglementation entrepris dans les années soixante-dix, aucune réduction notoire du taux de mortalité par cancer d’origine environnementale n’a pu être obtenue, même si l’on s’en tient aux évaluations les plus modérées. Les procédures de l’EPA sont conçues de telle façon que les extrapolations aux faibles doses sont réalisées de la même manière pour tous les produits chimiques quel que soit leur mécanisme cancérogène. Cette approche se démarque nettement de celle des autres agences gouvernementales. Comme nous l’avons mentionné, l’Union européenne et plusieurs gouvernements européens — l’Allemagne, le Danemark, la France, l’Italie, les Pays-Bas, le Royaume-Uni, la Suède, et la Suisse — font une distinction entre les cancérogènes qui sont génotoxiques et ceux qui ne le sont pas et envisagent l’évaluation du risque différemment dans l’un et l’autre cas. En général, les cancérogènes non génotoxiques sont traités comme des toxiques à seuil. Des seuils sans effet sont définis et des facteurs de sécurité sont appliqués pour assurer une grande marge de sécurité. Déterminer si un produit chimique doit être considéré ou non comme génotoxique doit faire l’objet d’un débat scientifique et requiert le jugement éclairé des experts.
La question fondamentale à laquelle il appartient de répondre est la suivante: quelle est la cause du cancer chez l’humain et quel est le rôle des cancérogènes environnementaux? Les facteurs héréditaires du cancer humain sont beaucoup plus importants qu’on ne le prévoyait initialement. Si l’on veut faire de réels progrès dans l’évaluation du risque cancérogène, il importe de mieux comprendre et les causes et les mécanismes du cancer. La recherche sur le cancer entre dans un champ d’investigation passionnant. La biologie moléculaire peut modifier radicalement nos conceptions sur les cancérogènes environnementaux, ainsi que la manière d’en assurer le contrôle et la prévention dans le milieu de travail comme dans l’environnement général. Il faut évaluer le risque cancérogène en étudiant les mécanismes d’action, qui sont des concepts totalement nouveaux, en particulier le mécanisme des cancers héréditaires et l’interaction des cancérogènes sur ce processus. Cette notion devra être prise en compte dans l’évaluation du risque des cancérogènes.